How was PKU discovered?
In 1934, two severely mentally retarded children were examined by Dr Asbjørn Følling. He proved, by classical organic chemistry, that they excreted phenylpyruvic acid in their urine. The substance was also found in the urine of eight additional mentally retarded patients.
What is the history of PKU?
Phenylketonuria (PKU) is an innborn error in phenylalanine metabolism. It was first described by Asbjörn Fölling in 1934 in Norway. PKU has been the paradigm of inherited metabolic disorders. It also allowed the proposition for the first biochemical explanation of mental retardation.
Who invented PKU?
Dr. Asbjörn Følling, a Norwegian biochemist and physician, first published the description of phenylketonuria (PKU) as a cause of mental retardation in 1934. Moreover, this study reported a laboratory test to confirm this metabolic disease, which was later determined to be an autosomal recessive metabolic disorder.
What does PKU mean?
Phenylketonuria (also called PKU) is a condition in which your body can't break down an amino acid called phenylalanine. Amino acids help build protein in your body. Without treatment, phenylalanine builds up in the blood and causes health problems.
What does PKU smell like?
One of the unique features of PKU is a “mousy” or “musty” odor to the skin, hair, sweat and urine due to the elevated phenylalanine levels.
Are PKU tests still done?
Although PKU is rare, all newborns in the United States are required to get a PKU test. The test is easy, with virtually no health risk. But it can save a baby from lifelong brain damage and/or other serious health problems.
When was the first PKU done?
In 1961, Robert Guthrie, a doctor and bacterial scientist at the University of Buffalo Children's Hospital, developed a way to test whether newborn babies have phenylketonuria (PKU), an inability to digest the amino acid phenylalanine.
What are other names for PKU?
Phenylketonuria (fen-ul-key-toe-NU-ree-uh), also called PKU, is a rare inherited disorder that causes an amino acid called phenylalanine to build up in the body.
When did they discover PKU?
Dr Asbjörn Fölling, the scientist, in mid-life, about the time of the discovery of PKU in 1934.
What is phenylalanine in Coke?
Some of our reduced and no sugar, no calorie drinks like Diet Coke and Coca‑Cola Zero Sugar are sweetened with aspartame, which contains a source of phenylalanine, and so all our products are carefully labelled to make this clear.
How long is the average lifespan of a person with PKU?
PKU does not shorten life expectancy, with or without treatment. Newborn screening for PKU is required in all 50 states. PKU is usually identified by newborn screening. A child's outlook is very good if she strictly follows the diet.
Can a person with PKU donate blood?
Yes! According to the World Health Organization (WHO) Blood Donor Selection Guidelines and the American Red Cross (800-Red-Cross) PKU is not an exclusion for blood donation. PKU is an inherited disorder and it cannot be transmitted by a blood transfusion.
When did they discover PKU?
Dr Asbjörn Fölling, the scientist, in mid-life, about the time of the discovery of PKU in 1934.
When was PKU first treated?
In remembering the scientific triumphs of the first case of dietary treatment for PKU in Birmingham in 1951, seventy years ago at the time of writing, we should also think about Sheila's family.
What year did PKU testing start?
In 1961, Robert Guthrie, a doctor and bacterial scientist at the University of Buffalo Children's Hospital, developed a way to test whether newborn babies have phenylketonuria (PKU), an inability to digest the amino acid phenylalanine.
How long have they been testing babies for PKU?
Accepted 2013 Sep 24. Just over 50 years ago, Dr Robert Guthrie developed a simple screening test for phenylketonuria (PKU) that became the prototype for universal newborn screening programs.
When did Fanconi read PKU?
Early one morning in 1949 Fanconi read a report on PKU and during morning rounds surprised Bickel by asking why their mentally deficient patients had not been screened with the ferric chloride test. Bickel had to admit he had never heard of PKU. They immediately began testing their patients for phenylpyruvic acid.
Who published the first successful trial of PKU dietary therapy?
Meanwhile, the two men who would publish the first successful trial of PKU dietary therapy, Horst Bickel and John Gerrard, were serving as medical officers on opposite sides of World War II—Gerrard with the British Army and Bickel with the German Navy.
Who discovered phenylpyruvic acid in urine?
A dozen years later British psychiatrist and geneticist Lionel Penrose observed that the test for phenylpyruvic acid in urine “is so simple and striking that the failure of clinicians to observe the reaction until so recently is puzzling, except on the basis of the difficulty of obtaining specimens from subjects of low mental grade.” Still, as Penrose added in his inaugural lecture at University College London, Følling’s discovery inspired a surge of urine testing of mentally deficient patients in institutions, with reports of findings published in succession in the United Kingdom, France, the United States, Switzerland, and Canada. A single neurophysiologist at a mental institution in New York State tested over 8,000 patients in the mid-1930s.
Who was the first person to identify phenylketonuria?
Følling was the first to identify the disease today known as phenylketonuria. Through her kitchen window Borgny Egeland surveyed grey skies made luminous by a midday sun hanging low on the horizon. The distant sound of the town hall’s chimes might have lightened her spirits, but Borgny had reached her wit’s end.
Who developed phenylalanine free formula?
The Hospital for Sick Children, now known as the Great Ormond Street Hospital, in 1910. Chemist L. I. Woolf, working at the hospital in the early 1950s, developed a phenylalanine-free formula for children with phenylketonuria. Great Ormond Street Hospital for Children NHS Trust.
Is PKU a disease?
If that theory were true, then PKU was an inherited defect of the liver, not the brain, and so might be treated with a phenylalanine-free diet. Granted, this disease was relatively rare, and a successful therapy would only help limited numbers of patients.
What is PKU in biology?
Phenylketonuria ( PKU) is an inborn error of metabolism that results in decreased metabolism of the amino acid phenylalanine. Untreated, PKU can lead to intellectual disability, seizures, behavioral problems, and mental disorders.
What is PKU genetics?
PKU is an autosomal recessive metabolic genetic disorder. As an autosomal recessive disorder, two PKU alleles are required for an individual to experience symptoms of the disease. For a child to inherit PKU, both the mother and father must have and pass on the defective gene. If both parents are carriers for PKU, there is a 25% chance any child they have will be born with the disorder, a 50% chance the child will be a carrier, and a 25% chance the child will neither develop nor be a carrier for the disease.
What is the pathophysiology of phenylketonuria?
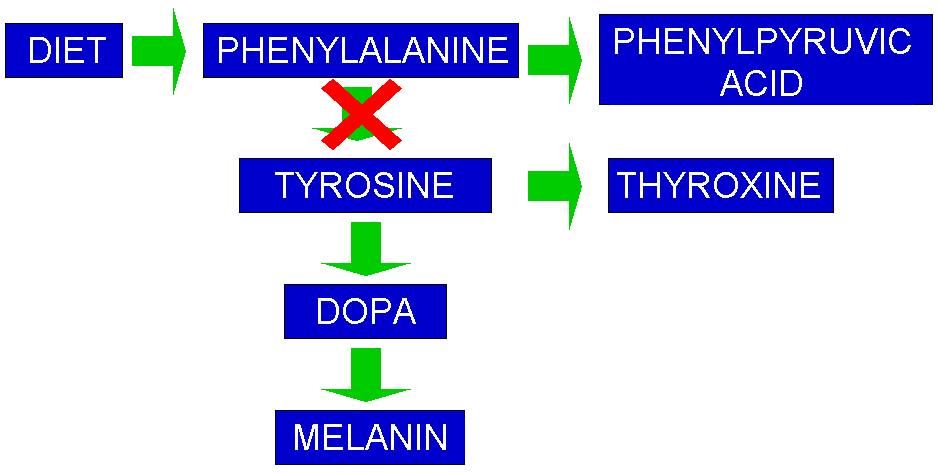
Pathophysiology of phenylketonuria, which is due to the absence of functional phenylalanine hydroxylase (classical subtype) or functional enzymes for the recycling of tetrahydrobiopterin (new variant subtype) utilized in the first step of the metabolic pathway.
What is phenylalanine PKU?
Sapropterin dihydrochloride, pegvaliase. Prognosis. Normal health with treatment. Frequency. ~1 in 12,000 newborns. Phenylketonuria ( PKU) is an inborn error of metabolism that results in decreased metabolism of the amino acid phenylalanine.
What are the effects of PKU?
Untreated, PKU can lead to intellectual disability, seizures, behavioral problems, and mental disorders. It may also result in a musty smell and lighter skin. A baby born to a mother who has poorly treated PKU may have heart problems, a small head, and low birth weight. Phenylketonuria is a genetic disorder inherited from a person's parents.
How many babies are affected by phenylketonuria?
Phenylketonuria affects about 1 in 12,000 babies. Males and females are affected equally. The disease was discovered in 1934 by Ivar Asbjørn Følling, with the importance of diet determined in 1953. Gene therapy, while promising, requires a great deal more study as of 2014.
How to treat phenylalanine in babies?
Babies should use a special formula with a small amount of breast milk. The diet should begin as soon as possible after birth and be continued for life.
When was PKU discovered?
The Discovery of PKU by Dr. Asbjørn Følling: Norway, 1934. This story is based on a talk given by Dr. Ivar Følling, son of the man who discovered PKU. It was presented at a meeting in Elsinore, Denmark, May 24-27,1994. The meeting was organized to celebrate the 60th anniversary of Dr. Asbjørn Følling’s discovery of PKU in Norway, 1934.
What is PKU in medicine?
PKU is obviously an inborn error of metabolism and it was the first to be shown to affect the mind. This was a completely new concept in medicine and to my mind this represents the major step. Today more than 200 such conditions are known and it is common to search for biochemical explanations in mental diseases.
What is the source of phenylpyruvic acid?
Loading experiments in animals and humans indicated that the high phenylalanine levels were probably the source of phenylpyruvic acid. The hypothesis of a block in phenylalanine metabolism was strengthened. They also found large amounts of phenyllactic acid in urine.
What was the ferric chloride used for in the urine of diabetics?
Urine examination showed no protein or glucose. He then added some ferric chloride to the urines. Ferric chloride was used to detect ketones in the urine of diabetics. Therefore this was part of his thorough routine examination.
Why is PKU considered a medical advance?
The discovery and elucidation of PKU is a medical advance because it has changed the lives of patients from one of disability to one of ability. However, to my mind there is a general aspect to the story which is more important and which represents the major step.
What is PKU in Norway?
The reason that I tell the story is that the discoverer was my father, and in Norway we often term it Følling’s disease, characteristic of the modesty of my countrymen.
Did Proteus miss the word "not"?
On rereading the article they realized that they had missed the word “not.”. Proteus was not able to do the conversion. Hence, their misreading had led to a fruitful discovery. Instead of measuring phenylpyruvic acid, they had actually measured phenylalanine.
Who discovered phenylpyruvic acid in children?
In 1934, two severely mentally retarded children were examined by Dr Asbjørn Følling. He proved, by classical organic chemistry, that they excreted phenylpyruvic acid in their urine. The substance was also found in the urine of eight additional mentally retarded patients. Based on these observations ….
What was the substance found in the urine of mentally retarded children?
In 1934, two severely mentally retarded children were examined by Dr Asbjørn Følling. He proved, by classical organic chemistry, that they excreted phenylpyruvic acid in their urine. The substance was also found in the urine of eight additional mentally retarded patients. Based on these observations, oligophrenia phenylpyrouvica (later termed ...
Who was the original King of Prussia?
It was named for its owner, a native of Prussia, in honor of Frederick I , who had, a short time before, establishing himself as the King of Prussia. To those who doubt there ever was a so-called “King”, the direct descendant of the original Frederick I , King of Prussia, is Louis Ferdinand, Prince of Prussia, visited Dr.
What road was King of Prussia's inn?
by Dennis Daylor, Courier Editor. …. The pre-revolutionary inn, located at the crossroads now known as Route 202 and Gulph Road, gave its name to King of Prussia in 1851. It served as a voting place of the community until well into the 20th century.

Simple and Striking
- Newborns with PKU initially don't have any symptoms. However, without treatment, babies usually develop signs of PKUwithin a few months. Signs and symptoms of untreated PKUcan be mild or severe and may include: 1. A musty odor in the breath, skin or urine, caused by too much phenyl…
A Cure For Mental Retardation?
“She Awaited Me Every Morning”
One Disease, Many Lessons
Overview
Signs and symptoms
Genetics
- As a metabolic expert, Følling immediately recognized that the phenylpyruvic acid in urine was likely connected to a metabolic problem. In his 1934 paper he speculated that PKU was caused by an inherited error in metabolizing an essential amino acid called phenylalanine, which had a chemical structure almost identical to that of phenylpyruvic acid....
Pathophysiology
- Ultimately, protein replacement would wait until the end of World War II. During the war years pioneers of PKU research like Følling and George Jervis worked on genetic and biochemical questions rather than therapeutic ones. They worked out the pattern of inheritance, elucidated the metabolic pathway of PKU and where it went awry, created and improved methods of chemical …
Screening
- Sheila wasn’t cured, but from these modest beginnings researchers would learn that if administered from birth, this dietary prescription would prevent mental disability. Using work on PKU as a model, support for research in the prevention and treatment of mental disability flourished. Screening programs were gradually established to identify at birth children with PKU, …
Treatment
Phenylketonuria (PKU) is an inborn error of metabolism that results in decreased metabolism of the amino acid phenylalanine. Untreated PKU can lead to intellectual disability, seizures, behavioral problems, and mental disorders. It may also result in a musty smell and lighter skin. A baby born to a mother who has poorly treated PKU may have heart problems, a small head, and low birth weight.
Phenylketonuria is a genetic disorder inherited from a person's parents. It is due to mutations in t…
Epidemiology
Untreated PKU can lead to intellectual disability, seizures, behavioral problems, and mental disorders. It may also result in a musty smell and lighter skin. A baby born to a mother who has poorly treated PKU may have heart problems, a small head, and low birth weight.
Because the mother's body is able to break down phenylalanine during pregnancy, infants with PKU are normal at birth. The disease is not detectable by physical examination at that time, bec…
History
PKU is an autosomal recessive metabolic genetic disorder. As an autosomal recessive disorder, two PKU alleles are required for an individual to experience symptoms of the disease. For a child to inherit PKU, both the mother and father must have and pass on the defective gene. If both parents are carriers for PKU, there is a 25% chance any child they have will be born with the disorder, a 50% chance the child will be a carrier and a 25% chance the child will neither develop nor be a ca…
The Discovery
When phenylalanine (Phe) cannot be metabolized by the body, a typical diet that would be healthy for people without PKU causes abnormally high levels of Phe to accumulate in the blood, which is toxic to the brain. If left untreated (and often even in treatment), complications of PKU include severe intellectual disability, brain function abnormalities, microcephaly, mood disorders, irregular motor functioning, and behavioral problems such as attention deficit hyperactivity disorder, as w…
Further Elucidation
PKU is commonly included in the newborn screening panel of many countries, with varied detection techniques. Most babies in developed countries are screened for PKU soon after birth. Screening for PKU is done with bacterial inhibition assay (Guthrie test), immunoassays using fluorometric or photometric detection, or amino acid measurement using tandem mass spectrometry (MS/MS). Measurements done using MS/MS determine the concentration of Phe a…
Perspective
PKU is not curable. However, if PKU is diagnosed early enough, an affected newborn can grow up with normal brain development by managing and controlling phenylalanine ("Phe") levels through diet, or a combination of diet and medication.
People who follow the prescribed dietary treatment from birth may (but not always) have no symptoms. Their PKU would be detectable only by a blood test. People must adhere to a specia…
Honor
The average number of new cases of PKU varies in different human populations. United States Caucasians are affected at a rate of 1 in 10,000. Turkey has the highest documented rate in the world, with 1 in 2,600 births, while countries such as Finland and Japan have extremely low rates with fewer than one case of PKU in 100,000 births. A 1987 study from Slovakia reports a Roma population with an extremely high incidence of PKU (one case in 40 births) due to extensive inbr…
Knowledge Leads to Humility
Before the causes of PKU were understood, PKU caused severe disability in most people who inherited the relevant mutations. Nobel and Pulitzer Prize winning author Pearl S. Buck had a daughter named Carol who lived with PKU before treatment was available, and wrote an account of its effects in a book called The Child Who Never Grew. Many untreated PKU patients born before widespread newborn screening are still alive, largely in dependent living homes/institutio…