How do you make a phylogenetic tree more accurate? In general, the more information you're able to compare, the more accurate the tree will be. So you'd get a more accurate tree by comparing entire skeletons, instead of just a single bone.
How do you make a phylogenetic tree from a DNA sequence?
So you’d get a more accurate tree by comparing entire skeletons, instead of just a single bone. Or by comparing entire genomes, instead of just a single gene. Any DNA, RNA, or protein sequence can be used to generate a phylogenetic tree. But DNA sequences are most commonly used in generating trees today.
What is the most accurate DNA phylogenetic tree?
However, a tree can be considered to be "the most accurate" if the same tree is obtained from different analysis, such as different gene regions, different loci, different DNA sequences and/or protein sequences. And different tree topologies, such MP, Ml, UPGMA, NJ, etc.
Do phylogenetic trees reflect true phylogenetic relationships?
Of course, a well-supported tree could fail to reflect the "true" phylogenetic relationships. This is the main concern of those worried about the gene tree-species tree problem, long branch attraction, etc.
How can we tell if a phylogeny is accurate?
These, in a phylogenetic context, are signal errors and systematic errors. With these errors in mind, we must admit that we can never tell if a phylogeny is accurate (except in special cases where we have made direct observations on the past, for example when culturing bacterial strains in the lab). All is not lost!
How can you increase the accuracy of a phylogenetic tree?
Our results imply that more accurate phylogenetic inference can be achieved by inclusion of larger numbers of taxa from lineages for which prior knowledge suggests that a change in the evolutionary process occurred.
How do you know if a phylogenetic tree is accurate?
The reliability of a phylogenetic tree obtained from empirical data is usually measured by the bootstrap probability (Pb) of interior branches of the tree. If the bootstrap probability is high for most branches, the tree is considered to be reliable.
What is the most accurate way to determine phylogenetic relationships?
The most generally applied method for determining phylogenetic relationships between microorganisms is based on comparative analysis of the 16S rRNA gene sequences (Neefs et al., 1990).
What is the most accurate phylogenetic tree?
Over the variety of conditions tested, Bayesian trees estimated from DNA sequences that had been aligned according to the alignment of the corresponding protein sequences were the most accurate, followed by Maximum Likelihood trees estimated from DNA sequences and Parsimony trees estimated from protein sequences.
What is the most reliable accurate method for inferring phylogenetic relationships?
Despite being slow and computationally expensive, maximum likelihood is the most commonly used phylogenetic method used in research papers, and it is ideal for phylogeny construction from sequence data.
What is the most common error in making and analyzing a phylogenetic tree?
With some DNA sequences - for example, the β-globin genes of different vertebrates - there is no difficulty in being sure that the sequences being compared are homologous, but this is not always the case, and one of the commonest errors that arises during phylogenetic analysis is the inadvertent inclusion of a non- ...
What is the best way to determine how closely organisms are closely related?
However, now scientists can also analyze DNA to discover how closely organisms are related. Every living creature has DNA, which has a lot of inherited information about how the body builds itself. Scientists can compare the DNA of two organisms; the more similar the DNA, the more closely related the organisms.
How do you know what is more closely related on a phylogenetic tree?
On a phylogenetic tree, more closely related terminal taxa are connected by shallower nodes (i.e., nodes nearer to the tips of the tree) and more distantly related terminal taxa are connected by deeper nodes (i.e., nodes nearer to the base of the tree).
What techniques are used for assessing robustness of phylogenetic trees?
Computer simulations provide a flexible method for assessing the power and robustness of phylogenetic inference methods.
What is used as evidence to develop a phylogenetic tree?
Any DNA, RNA, or protein sequences can be used to draw a phylogenetic tree. But DNA sequences are the most widely used.
What are the different methods for phylogenetic analysis?
Various methods including a molecular clock, midpoint rooting, and outgroup rooting, are available to accurately estimate the tree root using gene sequencing data and assumptions. In contrast, an unrooted phylogenetic tree only represents relationships among species without showing an ancestral root of origin.
How do you Analyse a phylogenetic tree?
In a phylogenetic tree, every leaf node represents a species, each edge denotes a relationship between two neighboring species and the length of an edge indicates the evolutionary distance among them.
How do you Analyse a phylogenetic tree?
In a phylogenetic tree, every leaf node represents a species, each edge denotes a relationship between two neighboring species and the length of an edge indicates the evolutionary distance among them.
Which of the following phylogenetic line is correct?
So, the correct option is 'Ontogeny repeats phylogeny'
Which of the following is true about phylogenetic trees?
Which of the following is true about phylogenetic trees? They're a way of visualizing how different species are related.
When creating phylogenetic trees What is the most exact information to utilize?
Any DNA, RNA, or protein sequences can be used to draw a phylogenetic tree. But DNA sequences are the most widely used. It's pretty cheap and easy now to get DNA sequences. Plus DNA contains more information, which can make more accurate trees.
Why do biologists use many different characteristics to build phylogenetic trees?
Biologists often use many different characteristics to build phylogenetic trees because of sources of error like these. Even when all of the characteristics are carefully chosen and analyzed, there is still the potential for some of them to lead to wrong conclusions (because we don't have complete information about events that happened in the past).
What would happen if we were biologists building a phylogenetic tree?
If we were biologists building a phylogenetic tree as part of our research, we would have to pick which set of organisms to arrange into a tree. We'd also have to choose which characteristics of those organisms to base our tree on (out of their many different physical, behavioral, and biochemical features).
What are some examples of traits that are shared by all species?
We see three new traits arising at different points during the evolutionary history of the group: a fuzzy tail, big ears, and whiskers . Each new trait is shared by all of the species descended from the ancestor in which the trait arose (shown by the tick marks), but absent from the species that split off before the trait appeared.
Why is the sea bass tree less parsimonious?
However, it is less parsimonious because it requires more independent changes in traits to take place. Because where we've put the sea bass, we have to hypothesize that jaws independently arose two separate times (once in the sea bass lineage, and once in the lineage leading to antelopes, bald eagles, and alligators). This gives the tree a total of tick marks, or trait change events, versus in the more parsimonious tree above.
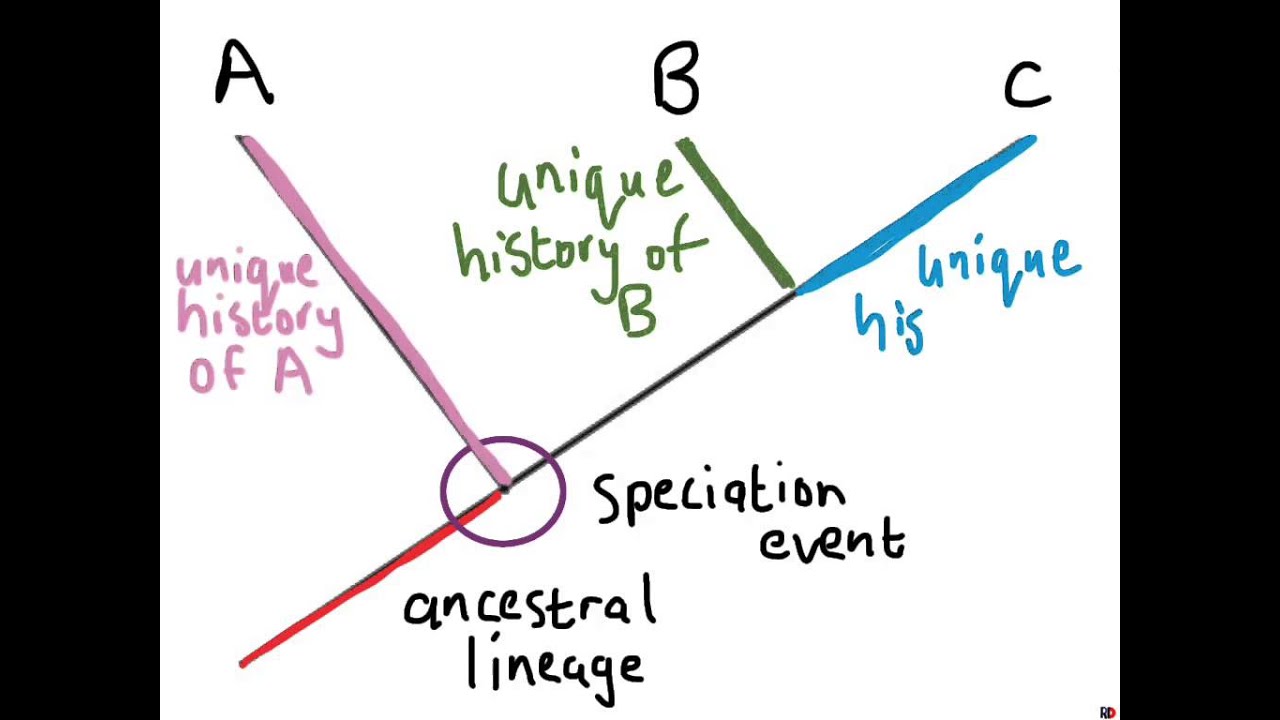
What is a phylogenetic tree?
Phylogenetic trees represent hypotheses about the evolutionary relationships among a group of organisms. A phylogenetic tree may be built using morphological (body shape), biochemical, behavioral, or molecular features of species or other groups. In building a tree, we organize species into nested groups based on shared derived traits ...
Which trait is shared by alligators and bald eagles?
Following the same pattern, we can now look for the derived trait shared by the next-largest number of organisms. That would be the gizzard, which is shared by the alligator and the bald eagle (and absent from the antelope). Based on this data, we can draw the antelope lineage branching off from the alligator and bald eagle lineage, and place the appearance of the gizzard on the latter.
How do phylogenetic trees connect?
In a phylogenetic tree, the species of interest are shown at the tips of the tree's branches . The branches themselves connect up in a way that represents the evolutionary history of the species—that is, how we think they evolved from a common ancestor through a series of divergence (splitting-in-two) events. At each branch point lies the most recent common ancestor shared by all of the species descended from that branch point. The lines of the tree represent long series of ancestors that extend from one species to the next.
Can you tell if a phylogeny is accurate?
Stopping short of the philosophical questions of what rationality is: - we cannot tell if a phylogeny is accurate, we can only tell if it is rational to believe that it is accurate.
Is a tree a good fit for a given evolutionary model?
At least you can say that a tree is a good fit to the data, for a given evolutionary model. Usually you can also say that certain trees provide a better fit to the data than others under almost any realistic model of character change. You can also say whether different datasets, or different partitions of a dataset support the same tree, and whether this is consistent with other information such as fossils or well dated historical events that would have impacted these taxa. Independent verification is the gold standard for assessing support of any hypothesis, isn't it?
Is a well supported tree true?
Of course, a well-supported tree could fail to reflect the "true" phylogenetic relationships. This is the main concern of those worried about the gene tree-species tree problem, long branch attraction, etc. In order to recognize these phenomena in the first place, one must have some sort of a priori idea about what the truth is - or at least a pattern of incongruent topologies from different data partitions. The former of these represents a correspondence theory of truth, which seems to be the position of people who talk about "realistic models" and that sort of thing (when you say something is "realistic," presumably you mean corresponding to reality in some way). The latter (congruence) represents a coherence theory of truth - "truth" is a confection of the agreement of evidence, and the more evidence that agrees, the more plausible the result is - but still no guarantee that it corresponds to the Kantian "thing-in-itself."
Can support metrics be used to assess the accuracy of a tree?
In a strict sense support metrics can only assess the precision or 'fit' of the data to the tree, evaluating the accuracy of any particular tree compared with the true evolutionary tree is not possible. Simulation studies can on the other hand provide some insight into how well tree search algorithms recover a correct topology with a artificial known dataset, and by extension known 'true' tree.
Is a phylogenetic tree accurate?
What my experience is that there is no "precise tree" in phylogenetic analysis. All you can obtain is a tree that supports most evidences and a tree that you think can support your hypothesis based on the morphoogical, distribution or whatever the character that you consider outside the molecular data. However, a tree can be considered to be "the most accurate" if the same tree is obtained from different analysis, such as different gene regions, different loci, different DNA sequences and/or protein sequences. And different tree topologies, such MP, Ml, UPGMA, NJ, etc.