What are the characteristics of dyslipidemia of insulin resistance?
The characteristic dyslipidemia of insulin resistance consists of elevated triglyceride and triglyceride-rich lipoprotein levels, low levels of high-density lipoprotein cholesterol, and increased concentrations of small, dense low-density lipoprotein cholesterol. Management of this dyslipidemia typically involves a dual approach.
How does insulin resistance affect triglycerides?
The result is more triglyceride-rich particles, fewer HDL particles, and more small, dense LDL. Postprandial triglyceride levels and measures of postprandial remnants also may contribute to increased coronary artery disease (CAD) risk in individuals with insulin resistance.
What is the relationship between diabetes dyslipidemia and high glucose levels?
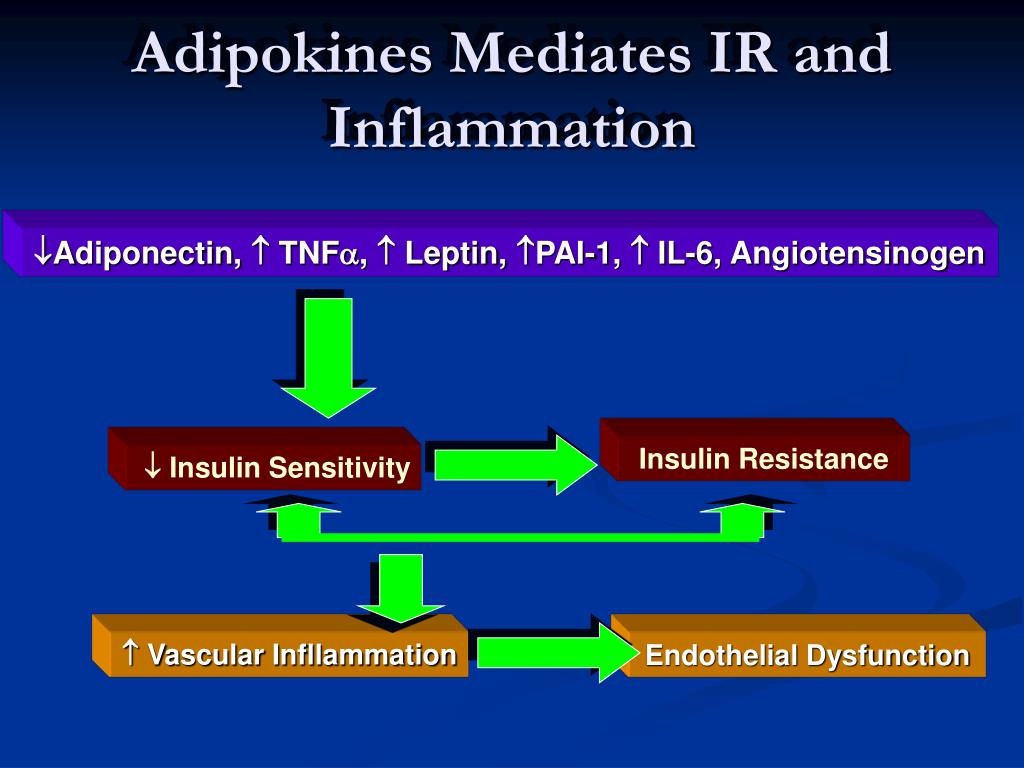
High glucose levels (hyperglycemia) and diabetic dyslipidemia do share a relationship, but it’s not the high glucose alone that causes abnormalities with the way your body handles lipids/fat, it’s insulin resistance. Insulin resistance occurs when the insulin receptor cells do not respond to the action of insulin on those cells.
How does insulin resistance affect the body?
The development of insulin resistance typically increases insulin production (hyperinsulinemia) so your body can maintain healthy blood sugar levels. Elevated levels of insulin can result in weight gain, which, in turn, makes insulin resistance worse. Hyperinsulinemia is also associated with the following conditions:

How does insulin resistance affect cholesterol?
Insulin resistance is associated with increased cholesterol synthesis, decreased cholesterol absorption and enhanced lipid response to statin therapy. Atherosclerosis.
How does insulin resistance affect lipids?
Insulin resistance in adipose tissue results in a reduction in the uptake of circulating free fatty acids and an increase in the hydrolysis of stored triglycerides by lipases. The net result of these actions is an increase in circulating free fatty acids.
Why does diabetes cause dyslipidemia?
Several factors are likely to be responsible for diabetic dyslipidemia: insulin effects on liver apoprotein production, regulation of lipoprotein lipase (LpL), actions of cholesteryl ester transfer protein (CETP), and peripheral actions of insulin on adipose and muscle.
How does insulin resistance lead to atherosclerosis?
Insulin resistance also decreases lipoprotein lipase activity, the major mediator of VLDL clearance, which may make a smaller contribution to elevated triglycerides in this setting. VLDL is metabolized to remnant lipoproteins and LDL, both strongly associated with atherosclerotic risk.
Why does insulin resistance cause low HDL?
Compensatory hyperinsulinemia due to IR can induce increased flux of free fatty acids, raise production of triglycerides in the liver, and decrease high-density lipoprotein cholesterol (HDL-C).
Does insulin resistance cause lipolysis?
In general, insulin resistance leads to increased levels of lipolysis, and improvement in insulin sensitivity inhibits excessive lipolysis in adipose tissue.
How are hyperglycemia and dyslipidemia linked?
Glucagon becomes the major hormone controlling blood glucose, which leads to a mobilization of fats into the blood to be manufactured into glucose. This leads to both elevated blood glucose (hyperglycemia) and elevated plasma lipids (lipidemia).
What is the relationship between hyperlipidemia and diabetes?
Hyperlipidemia is common in patients with diabetes mellitus and is partly responsible for the increased vascular disease seen in these patients. Effective drugs are now available for its treatment. More attention must be given to hypertriglyceridemia and reduced HDL-C levels in such patients.
What is the main cause of dyslipidemia?
The most common conditions that were felt to be contributing to dyslipidemia were excessive alcohol intake (10 percent) and uncontrolled diabetes mellitus (8 percent). Hyperlipidemia in association with insulin resistance is common in patients with type 2 diabetes mellitus [2,3].
How does insulin affect arteries?
And what insulin does in our arteries sends a signal that helps prevent the buildup of fatty plaques that can cause arteries to harden, new research in mice shows. Earlier studies showed that in the context of systemic insulin resistance, blood vessels become resistant, too.
What is the relationship between atherosclerosis and diabetes?
Both types of diabetes mellitus have been shown to be independent risk factors for accelerated atherosclerosis development. It is now clear that the pathogenesis of diabetes mellitus and atherosclerosis are closely linked, but the mechanisms and molecular interactions of this linkage are still under discussion.
Why does diabetes cause atherosclerosis?
Scientists have known that diabetes leads to atherosclerosis. The study shows that this is associated with increased circulating levels of inflammatory white blood cells (WBCs), which contribute to the build-up of plaque.
What effect does insulin have on blood lipids?
In addition to glucose regulation, insulin also controls blood lipid levels in part by increasing lipid synthesis, uptake, and storage in fat cells.
Does insulin resistance increase fat?
If your cells become too resistant to insulin, it can result in elevated blood sugar levels, which can lead to weight gain, prediabetes and type 2 diabetes. Losing weight with insulin resistance is more difficult because the body stores excess blood sugar as fat.
Does insulin regulate lipids?
Insulin Regulates Hepatic Triglyceride Secretion and Lipid Content via Signaling in the Brain.
Does insulin increase lipid synthesis?
Insulin therapy stimulates lipid synthesis and improves endocrine functions of adipocytes in dietary obese C57BL/6 mice - PMC. The .
Insulin resistance and dyslipidemia
Ronald Krauss (San Francisco, CA) gave insights in the genetics of the more common forms of IRS based on the understanding of the complex topic of dyslipidemia. There is an intimate relationship between diet and dyslipidemia, and dietary manipulations may be used to understand the mechanisms of these abnormalities.
Cardiovascular disease and insulin resistance
Andrew Selwyn (Boston, MA) reviewed aspects of the relationship between IRS and CVD, noting the increasing prevalence of type 2 diabetes from the 1970s to the 1990s, with its far stronger association with CVD than with the traditional microvascular complications such as visual loss, renal failure, and amputation ( 10 ).
How does fructose affect the liver?
Hepatic fructose metabolism: A highly lipogenic pathway. Fructose is readily absorbed from the diet and rapidly metabolized principally in the liver. Fructose can provide carbon atoms for both the glycerol and the acyl portions of triglyceride. Fructose is thus a highly efficient inducer of de novo lipogenesis. High concentrations of fructose can serve as a relatively unregulated source of acetyl CoA. In contrast to glucose, dietary fructose does NOT stimulate insulin or leptin (which are both important regulators of energy intake and body adiposity). Stimulated triglyceride synthesis is likely to lead to hepatic accumulation of triglyceride, which has been shown to reduce hepatic insulin sensitivity, as well as increased formation of VLDL particles due to higher substrate availability, increased apoB stability, and higher MTP, the critical factor in VLDL assembly.
What are the causes of insulin resistance?
The main driving forces for the increased prevalence of insulin resistance are modern Westernized diets and patterns of eating associated with the dramatic rises in obesity. Insulin resistance is often linked to the macronutrient content in the diet. In the past, diets high in saturated fats have been shown to induce weight gain, insulin resistance, and hyperlipidemia in humans and animals [19-22]. Recent research suggests that a high intake of refined carbohydrates may also increase the risk of insulin resistance [23-26]. In addition, diets specifically high in fructose have been shown to contribute to a metabolic disturbance in animal models resulting in weight gain, hyperlipidemia [27], and hypertension [28].
What is the glycemic index?
The glycemic index (GI) has been commonly used to differentiate and compare various nutrients, as well as to describe how different foods produce different plasma glucose levels after ingestion. The GI can range from 100 for glucose and baked potato compared to approximately 20 for fructose and whole barley [54]. Foods with varying GIs have different time courses associated with satiety. High GI carbohydrates have been reported to reduce appetite in the short term, whereas low GI carbohydrates possess a more delayed effect on energy intake controls [55]. Fructose appears to have differing effects on appetite compared to glucose, contributing to its negative properties. Anderson et al. determined the association between food intake and blood glucose, comparing glucose and a fructose mixture. Glucose was administered as a high GI preload, which resulted in lower mealtime energy intakes compared to the low GI preload of the glucose-fructose mixture. An inverse relationship was seen between GI (and blood glucose concentrations), and appetite with consequent increased food intakes seen with fructose [56]. In 2002, Vozzo et al. studied the comparative effects of glucose and fructose on blood glucose, insulin, and acute food intake. When subjects drank equienergetic preloads of glucose or fructose before an ad libidum buffet lunch, glucose concentrations were lower in the fructose group compared to glucose, and insulin concentrations were 50% higher in the fructose group in type 2 diabetics than in non-diabetics. The authors concluded that fructose may be a suitable replacement for glucose in diabetic patients – although it was found that satiating efficiencies of fructose certainly offered no advantages [57]. This study differs from others with regards to insulin secretion, but the trend is clear between GI, glucose concentrations, and appetite. An explanation for the variation in glucose and fructose glycemic responses appears to be dependent on rates of hydrolysis and absorption of glucose, and gastric emptying [58]. The variations observed in GI and appetite control of glucose and fructose can also be explained by differences in stimulation of insulin and leptin, important players in the long-term regulation of energy homeostasis. Fructose will generally produce smaller insulin excursions upon consumption because it does not stimulate the secretion of insulin from pancreatic beta cells, whereas glucose does. Insulin-regulated leptin will also have a reduced concentration and a decreased net effect on reducing appetite. Limited effects on appetite suppression, combined with the fact that fructose is favoured by the liver to be metabolized into lipid, will subsequently lead to weight gain, hyperinsulinemia, and the associated insulin resistance [59]. Glucose and fructose comparison studies continued examining new hormonal targets. In 2004, Teff et al. showed that subjects served meals with either 30% glucose beverages, or 30% fructose beverages, had differing hormonal and metabolic responses. Glycemic excursions and insulin responses were reduced by 66% and 65%, respectively, in the fructose-consuming subjects. There was a concomitant reduction in circulating leptin both in the short and long-term as well as a 30% reduction in ghrelin (an orexigenic gastroenteric hormone) in the fructose group compared to the glucose group. A prolonged elevation of TG was also seen in the high fructose subjects [60]. Both fat and fructose consumption usually results in low leptin concentrations which, in turn, leads to overeating in populations consuming energy from these particular macronutrients. An adipocyte hormone, adiponectin, also plays an important role in lipid homeostasis and insulin action [61]. The insulin sensitizer agonist, peroxisome proliferator-activated receptor-gamma, stimulates adiponectin production and adiponectin is in fact thought to be part of this agonist's mechanism lowering circulating fatty acids and increasing fat oxidation. The net effect is to decrease liver TG and increase insulin sensitivity [62]. Chronic fructose consumption reduces adiponectin responses, contributing to insulin resistance [63].
What is metabolic syndrome?
The metabolic syndrome, also referred to as "Diabesity" [1] describes the increasing incidence of diabetes in combination with obesity as a result of changes in human behaviour, available nutrition, and the adoption of more sedentary lifestyles. Obesity and type 2 diabetes are occurring at epidemic rates in the United States [2-4] and developing countries including China [5] and India [6]. From 1935 to 1996, the prevalence of diagnosed type 2 diabetes climbed nearly 765% [7]. The global figures are predicted to rise 46% from 150 million cases in 2000 to 221 million in 2010 [8]. This epidemic of type 2 diabetes is complicated by the fact that it is a multi-factorial disease, frequently associated with a cluster of pathologies including obesity, hypertriglyceridemia, impaired glucose tolerance, and insulin resistance, collectively referred to as the metabolic syndrome (formerly known as syndrome X and insulin resistance syndrome). Although there is no universally accepted definition of the metabolic syndrome, most would agree that the syndrome includes a cluster of common pathologies: obesity, insulin resistance, dyslipidemia, and hypertension. It is present in 25–50% of the United States population [9]. There has been a heightened awareness of the metabolic syndrome and a subsequent increase in clinical attention directed towards prevention, due to its strong association with premature morbidity and mortality [8,10]. In particular, these risk factors predispose the individual to greater risk for developing cardiovascular disease and Type 2 diabetes. Recently, the National Cholesterol Education Panel (NCEP) has officially described and identified a number of these risk factors for cardiovascular diseases [11]. These include: 1) abdominal obesity, 2) elevated TG levels, 3) low high density lipoprotein (HDL)-cholesterol levels, 4) increased blood pressure, and 5) impaired fasting glucose [12]. There is also now consensus that insulin resistance and obesity are actually part of one common pathologic mechanism of the metabolic syndrome [13,14]. Evidence shows that the metabolic syndrome process begins early in life and persistence from childhood to adolescent/adult life produces type 2 diabetes and cardiovascular disease [15,16]. The symptoms of metabolic syndrome are not necessarily manifestations of age, but develop over a predisposed background established at a young age [17,18]. This is a dangerous predisposition, with trends in modern diet and habit likely influencing health and behaviour in increasingly younger populations.
Does fructose affect lipogenesis?
Observations made in our own laboratory have also shown aberrant lipogenesis activity. In primary hepatocytes isolated from fructose fed hamsters, there were significant increases in LXRα, SREBP-1, FAS and SCD, which indicate increased activity of the lipogenic pathways (unpublished observations). Fructose has also been implicated in reducing PPARα levels in rat hepatocytes. PPARα is a ligand activated nuclear hormone receptor that is responsible for inducing mitochondrial and peroxisomal β-oxidation. Nagai et al. found that following 8 weeks of a high fructose diet, rats showed decreased PPARα mRNA and protein levels [88]. In addition, primary rat hepatocytes treated with fructose also showed decreased PPARα expression, suggesting that fructose or its metabolites can directly regulate lipid oxidation. We have also recently detected decreased mRNA levels of PPARα in both liver and intestine of the fructose fed hamster (unpublished observations). Hence, decreased PPARα expression can result in reduced oxidation, leading to cellular lipid accumulation. For example, PPARα null mice have extensive hepatic steatosis because of diminished β-oxidation capacity, such as seen in the insulin resistant state [113]. Other mechanisms have been illustrated by Taghibiglou et al., who found evidence for enhanced lipoprotein assembly, reduced intracellular apoB degradation, and increased microsomal triglyceride transfer protein (MTP) mass, mRNA and activity in the fructose fed hamster [100]. These metabolic changes also coincided with a decrease in ER-60, a cysteine protease that may play a role in apoB degradation, and an increase in synthesis and secretion of apoB [101]. It appears that a complex relationship exists in the fructose fed animal model that links insulin resistance and dyslipidemia through NEFA flux, SREBP-1 expression, de novo lipogenesis and MTP expression. Amplified MTP activity and expression would be expected to stimulate the assembly and secretion of apoB-lipoproteins, as an association has been demonstrated between MTP levels and VLDL production [114]. As insulin is a negative regulator of MTP gene expression [115], the upregulation of MTP that has been observed in insulin resistance states is predictable. MTP is also negatively regulated by SREBP through sterol response element (SRE) regions located within -124 and -116 of the 5' MTP gene promoter [116]. However, in fructose fed animals [87] as well as other models of insulin resistance [117] where increased levels of MTP and SREBP have been established, the regulatory effects of SREBP may play a minor role in regulating MTP expression. Rather increased hepatic NEFA and increased TG stores might stimulate MTP expression [118]. Recent observations in our laboratory show that oleic acid can stimulate the MTP promoter and the stimulation occurs independently of SRE activity (unpublished observations). Thus, in insulin resistance states, increased MTP may occur through another mechanism that may block SREBP-mediated inhibition of the promoter. These phenomena help explain the increased assembly and secretion of apoB in fructose fed models. In addition, increased levels of small dense LDL particles have been observed in insulin resistant states [119]. Early studies by Verschoor et al. showed that fructose diets altered the structure and function of VLDL particles causing and increase in the TG: protein ratio, and an increased total cholesterol and phospholipid content [120]. LDL particle size has been found to be inversely related to TG concentration [121] and therefore the higher TG results in a smaller, denser, more atherogenic LDL particle, which contributes to the morbidity of the metabolic disorders associated with insulin resistance. Several theories are proposed for the overproduction of VLDL: more TG per VLDL particle, increases in particle number, changes in the production rates of VLDL TG or apoB, decreased TG clearance, increased lipoprotein lipase activity, and increased de novo lipogenesis. It is likely a combination of some or all of these factors that contribute to the elevated TG seen in a fructose rich carbohydrate fed model of metabolic disorder. High fructose, which stimulates VLDL secretion, may initiate the cycle that results in metabolic syndrome long before type 2 diabetes and obesity develop [103].
Does fructose cause VLDL to increase?
Another contributing factor to VLDL overproduction includes fructose effects on lipid peroxidation. High fructose diets can have a hypertriglyceridemic and pro-oxidant effect, and fructose fed rats have shown less protection from lipid peroxidation. Replacing the fructose in these diets with a more natural source of high fructose, honey, reduces this susceptibility and lowers plasma nitrite and nitrate levels [109]. In 2004, Kelley et al. hypothesized that pro-oxidant stress response pathways may mediate hepatic increases in VLDL secretion and delayed clearance upon fructose feeding. Hypertriglyceridemic fructose fed rats were treated with lipoxygenase inhibitors, which reversed the inflammatory protein activity response, and the lipid dysregulation observed [102]. Recent findings have also shown that the hyperlipidemic and pro-oxidant effect induced by a high fructose diet can be decreased by oligofructose consumption. Oligofructose administered to fructose fed rats did not alter insulin concentrations, and lowered plasma leptin by 50% compared to control groups. Oligofructose prevented TG changes induced by fructose feeding, and decreased hepatic TG accumulation. The peroxidation effect of fructose was also decreased by oligofructose, and had beneficial protective effects [110]. Oxidative stress has often been implicated in the pathology of insulin resistance induced by fructose feeding, and lipid peroxides, diene conjugates, and reactive substances are undeniably elevated in fructose fed animals, especially accompanying a deficient antioxidant system. Administration of alpha-lipoic acid (LA) has been shown to prevent these changes, and improve insulin sensitivity [111]. LA treatment also prevents several deleterious effects of fructose feeding: the increases in cholesterol, TG, activity of lipogenic enzymes, and VLDL secretion, the reductions in lipoprotein lipase and HDL cholesterol and may even normalize a dyslipidemic cholesterol distribution of plasma lipoproteins [112]. Taken together, this evidence shows a clear role of peroxidative stress pathways involved in VLDL oversecretion.
Does fructose cause insulin resistance?
There is growing evidence that the insulin resistant state developed upon fructose feeding is also associated with stimulated hepatic VLDL secretion. Several animal models have been employed to examine the mechanisms of this induction of VLDL, and the subsequent increases in plasma TG observed. Mechanistic studies based on carbohydrate versus lipid metabolism have recently become important because carbohydrate induced hypertriglyceridemia shares a metabolic basis with high fat diet induced endogenous hypertriglycerolemia. The similarly induced dyslipidemias would therefore have the same or similar atherogenic risks [97]. Carbohydrate induced hypertriglycerolemia results from a combination of both TG overproduction, and inadequate TG clearance [97,98]. These disease processes and the hepatic steatosis caused by stimulated lipogenesis have been illustrated by fructose fed animal models showing how aberrant leptin signaling, hyperinsulinemia, and dyslipidemia are related to TG induction [95]. Animals maintained on a chronic high fructose diet develop elevated NEFA and hyperinsulinemia at the expense of glycemic control [99]. This is not surprising, as fructose-induced metabolic dyslipidemia is usually accompanied by whole body insulin resistance [100] and reduced hepatic insulin sensitivity [101]. In the fructose fed hamster model, animals showed decreased glucose disappearance rates, increased plasma NEFA and increased plasma and liver TG [27]. Figure Figure11(adapted from ref. 100) shows clear in vivo evidence of fructose-induced insulin resistance as assessed by euglycemic hyperinsulinemic clamp studies. Taghibiglou et al. further characterized the fructose fed hamster model demonstrating the development of a metabolic dyslipidemic state characterized by high plasma levels of VLDL-TG and apolipoprotein B (apoB) due to hepatic lipoprotein overproduction [100]. Serum TGs are elevated via both an increased secretion, and decreased clearance of VLDL [102]. Also, high rates of lipolysis in visceral adipose depots can increase availability of NEFAs and promote hepatic TG synthesis. The TG is then packaged with apoB, and secreted as VLDL particles [93]. Evidence has shown that there is a complex interplay of cellular enzymes regulating lipid synthesis and uptake, as well as export and oxidation. Observations of the actions of insulin affecting lipid secretion as well as inhibition of TG has brought research interests towards the effects of chronic insulin stimulation on VLDL secretion and transport. Excess VLDL secretion has been shown to deliver increased fatty acids and TG to muscle and other tissues, further inducing insulin resistance [103]. Induced cellular changes include alterations in hepatic pyruvate dehydrogenase, changes in insulin signaling phosphorylation, and increases of inflammatory cytokines [104,105]. It is evident that the metabolic effects of fructose occur through rapid utilization in the liver due to the bypassing of the regulatory phosphofructokinase step in glycolysis. This in turn causes activation of pyruvate dehydrogenase, and subsequent modifications favoring esterification of fatty acids, again leading to increased VLDL secretion [53]. Increases in VLDL secretion can then lead to chain reactions in other lipoproteins and lipids, such as low density lipoprotein (LDL). Resultant LDL cholesterol levels induced by high fructose intake are illustrated by comparison of a diet including 20% fructose, contrasted to a starch diet of less than 3% fructose. The 20% fructose diet initiated a cycle of increased fasting serum total and LDL cholesterol of 9% and 11%, respectively, over the starch feeding [106]. Increased evidence was shown in transgenic apo AI-CIII-AIV mice, fed a fructose solution for 9 months, where differential expressions of the apo AI and apo AIV genes were found. This indicated general perturbations in response to dietary intakes, causing long-term adverse effects in this hyperlipidemia mouse model [107]. The male Wistar fatty rat model of obese type 2 diabetes has also shown hyperglycemia. Remarkably, the female Wistar rats only develop this hyperglycemia when given sucrose, containing the responsible element of fructose, which causes increases in gluconeogenic enzymes and decreases in glucokinase. A hypertriglyceridemic effect is seen, presumably due to hepatic overproductions of VLDL and induction of lipogenic enzymes via dietary fructose [108].
What causes VLDL-TG removal?
VLDL-TG removal is impaired in patients with type 2 diabetes, which promotes hypertriglyceridemia accompanied by an increase in secretion of VLDL. The removal defect is mainly caused by reduced activity of LPL, particularly in adipose tissue13). Since insulin is an activator of LPL, insulin deficiency or insulin resistance diminishes LPL activity. In addition, increased serum levels of apoC3 could also contribute to the decreased VLDL catabolism52, 53). ApoC3 deficient mice were hypotriglyceridemic by enhanced VLDL-TG removal, and never developed hypertriglyceridemia even if obesity54)or diabetes55)were induced. Insulin suppresses apoC3 expression in the liver through downregulation of FOXO156). Insulin resistance conversely stimulates apoC3 production by FOXO1 activation. ApoA557)and angiopoietin-like proteins58)play an important role in regulating LPL activity and thereby TRL catabolism. It remains largely unknown how insulin/insulin resistance affects these key molecules affecting TRL catabolism. Table 1lists the comparison of insulin versus insulin resistance on major metabolic abnormalities of VLDLs.
What is the most common lipid abnormality in diabetics?
Hypertriglyceridemia is the most common serum lipid abnormality in diabetic populations. Serum TG levels are not simply elevated along with the degree of hyperglycemia, but hyperinsulinemia compensated by insulin resistance is closely correlated with TG levels4). TG consists of three molecules of fatty acids, the availability of circulating fatty acids (free fatty acids (FFA)) plays a crucial role in TG production in the liver5, 6), and partly in the intestine6, 7). Reaven et al.1)proposed that three distinct syndromes of hypertriglyceridemia occur as a result of abnormalities of glucose metabolism. In patients with impaired glucose tolerance, the basic defect is postulated to be the loss of normal insulin sensitivity, leading to compensatory hyperinsulinemia increased VLDL-TG secretion. Patients with type 2 diabetes have relative insulin deficiency, and the elevated FFA levels increase hepatic VLDL-TG secretion. In absolute insulin-deficient patients with type 1 diabetes, however, elevated FFA levels do not stimulate hepatic VLDL-TG secretion because the livers cannot respond to the increased FFA flux under severe insulin deficiency. The hypertriglyceridemia in patients with type 1 diabetes is primarily due to defect in the removal of VLDL-TG. This proposal provides important fundamental knowledge for understanding diabetic hypertriglyceridemia. However, one point remains questionable: Does hyperinsulinemia increase hepatic VLDL production? I will discuss this important issue later.
What is the role of TRLs in diabetes?
Production of VLDL and chylomicrons (TG-rich lipoproteins (TRLs)) is stimulated in individuals with type 2 diabetes. The long residence time of TRLs in circulation promotes excessive transfer of TG to LDL and a concomitant transfer of cholesteryl esters (CE) to TRLs via the action of cholesteryl ester transfer protein (CETP). Hepatic TG lipase-mediated hydrolysis of core TG produces cholesterol-poor LDL particles (small dense LDL).
What is pioglitazone used for?
Pioglitazone, a peroxisome proliferator-activated receptor (PPAR) gamma agonist, is an agent for the amelioration of insulin resistance , which might be an appropriate tool to study the relationship between insulin resistance and lipid metabolism . Pioglitazone has shown to improve TG and HDL-C levels and favorable effects on LDL particle size59). We examined the effect of pioglitazone on VLDL-TG metabolism in rats with severe insulin resistance60). Pioglitazone normalized TG levels by enhancing VLDL-TG removal from the circulation. Pioglitazone activated serum LPL activity and LPL production in adipose tissues in insulin-resistant mice61). Unexpectedly, pioglitazone did not suppress VLDL-TG overproduction. Nagashima et al.62)reported the same results in patients with type 2 diabetes. They found that apoC3 production was suppressed by pioglitazone, which resulted in increasing LPL activity. These results suggest that this insulin sensitizer does not necessarily ameliorate all abnormal lipid metabolism associated with insulin resistance.
Where are chylomicrons produced?
Chylomicrons are temporarily produced in the small intestine after dietary fat ingestion. Abnormal increase in serum TG levels after meal is called postprandial hyperlipidemia8), which is often observed in diabetic patients and reported to be associated with the incidence of CVD9). Chylomicron production is not only influenced by the amount of dietary fat but also by insulin resistance on enterocytes10). Similar to VLDL production in the liver, chylomicron production in the intestine is regulated by insulin and circulating FFA11). Lipoprotein lipase (LPL), which is attached to the luminal surface of vascular endothelium plays a major role in chylomicron removal by hydrolysing chylomicron-TGs12). LPL is upregulated by insulin, and conversely, insulin resistance diminishes LPL activity13). Severe chylomicronemia (so-called diabetic lipemia14)) occasionally occurs in patients with insulin-depleted diabetes due to LPL deficiency. In type 2 diabetes with insulin resistance, chylomicron levels are often elevated by both overproduction and catabolic defect of chylomicrons6, 7). Increased secretion of apoC3 (an inhibitor of LPL) into plasma contributes to less efficient lipolysis of chylomicron-TGs15). Although about 80% of the increase in TGs after a fat-load meal comes from chylomicrons16), approximately 80% of the increase in particle number is accounted for by VLDL particles containing apoB10017)Chylomicrons and VLDL particles are cleared from the circulation by common pathways and, therefore, compete for clearance18). For this reason, increased secretion of VLDL by the liver is an important predictor of postprandial accumulation of chylomicrons19).
Is LDL cholesterol negatively regulated?
LDL size is negatively regulated by serum TG levels, and LDL size is significantly reduced in hypertriglyceridemic subjects. However, individuals with severe hypertriglyceridemia, such as chylomicronemia, have only modest increase in CVD risk75). This suggests that measurement of LDL particle size is not sufficient to predict CVD, but the number of LDL particles should be taken account for evaluating the overall risk. We established a direct method for the quantification of sdLDL-C, which is applicable to the auto-analyzer76). This method quantifies only cholesterol in LDL particles of density between 1.044 and 1.063 g/L. Our assay has been employed in many large cohort studies77, 78)and has revealed that sdLDL-C is a potent predictor of CVD beyond LDL-C. SdLDL-C levels are increased with either levels of TG or LDL-C, and remarkably increased in the combined hyperlipidemia (Type IIb)79). As shown in Fig. 3, sdLDL-C levels corresponding to LDL-C levels were constantly higher in patients with diabetes or coronary artery diseases than in healthy controls80). SdLDL-C levels are highly (r= 0.94) correlated with sdLDL-apoB levels81), suggesting that measuring sdLDL-C is sufficient to estimate the number of sdLDL particles. The preponderance of sdLDL particle would be an essential mechanism for explaining a high incidence of CVD in diabetes.
Is LDL-C a predictor of CVD?
Unlike nephrotic syndrome or hypothyroidism, diabetes is not a representative disease exhibiting severe hypercholesterolemia. However, LDL-C is the best predictor of CVD66), and LDL lowering by statin treatments dramatically reduced the incidence of CVD in diabetes populations67). Why do LDL-C concentrations strongly affect CVD outcomes in diabetic patients. This is because the nature of LDL particles in diabetic patients are changed to more atherogenic than those in nondiabetic patients. LDL particles are not created equally68). Austin and Krauss discovered that individuals with small sized LDL particles (pattern B) had substantially higher prevalence of CVD than those with large-buoyant LDL particles (pattern A) determined by the gel electrophoresis69). SdLDL particles are thought to be more atherogenic than large-buoyant LDL particles as a result of their better penetration of the arterial wall, lower binding affinity for the LDL receptor, longer plasma half-life, and weaker resistance to oxidative stress70). Several studies have reported a 2- to 3-fold increase in the risk of CVD in patients with pattern B LDL particles69, 71). LDL size became smaller, and the incidence of the pattern B were higher in diabetic patients70). The formation of sdLDL is closely associated with insulin resistance and hypertriglyceridemia70), and VLDL1-TG level is the major predictor of LDL size48, 51, 70). The mechanisms for formation of sdLDL are estimated as follows: 1) Cholesteryl ester transfer protein (CETP) facilitates the transfer of TGs from TRLs to LDL, 2) the resulting TG-rich LDL is a preferred substrate for hepatic lipase; and 3) increased lipolysis of TG-rich LDL results in the formation of cholesterol-poor LDL, that is, sdLDL72)(Fig. 2). In metabolic syndrome, LDL-C levels are usually normal, but LDL particle size becomes small and the number of LDL particles is increased73). Abdominal fat volume measured by CT scan was positively associated with sdLDL-C but inversely associated with largebuoyant LDL-C74). Therefore, we can call sdLDL as “Metabolic LDL.”
What is Diabetic Dyslipidemia?
Diabetic dyslipidemia is a term that refers to alteration of blood lipids, or in other words, changes to your cholesterol levels. See this info for normal cholesterol levels.
Why is dyslipidemia a concern?
So the reason that diabetic dyslipidemia is a major concern is because it puts you at even greater risk for cardiovascular disease. If you have cholesterol abnormalities, you should pay attention and get proactive so you can reduce your risk of heart attack and stroke. Keep reading to learn more….
What is the relationship between high glucose and dyslipidemia?
High glucose levels (hyperglycemia) and diabetic dyslipidemia do share a relationship, but it’s not the high glucose alone that causes abnormalities with the way your body handles lipids/fat, it’s insulin resistance. Insulin resistance occurs when the insulin receptor cells do not respond to the action of insulin on those cells.
Why does insulin resistance affect blood glucose?
But it’s not just blood glucose that’s affected because insulin resistance influences lipid/fat metabolism too.
What are the factors that increase the risk of cardiovascular disease?
Increased small dense LDL particles. These are all factors that put you at increased risk of cardiovascular disease. However, the most important part of the equation, is the small dense LDL particles. For many years it’s been thought that having raised LDL cholesterol was the biggest issue.
What is the term for a person with type 2 diabetes?
You may have come across the term ‘diabetic dyslipidemia’ at the doctor’s office or when browsing the web. Diabetic dyslipidemia is a term that refers to alteration of blood lipids, or in other words, changes to your cholesterol levels. In people with type 2 diabetes, dyslipidemia can be an issue in 35 to 56% of people.
How to lower cholesterol?
Eat more fiber. Fiber has been shown time and time again to help reduce cholesterol levels, particularly soluble fiber. And chances are, you’re probably not getting enough fiber. As you cut down on pasta, potatoes, rice, noodles and bread, fill your plate with more non-starchy veggies.