What is a sickle cell crisis?
What is a sickle cell crisis? A sickle cell crisis is pain that can begin suddenly and last several hours to several days. It happens when sickled red blood cells block small blood vessels that carry blood to your bones. You might have pain in your back, knees, legs, arms, chest or stomach.
What are five symptoms of a sickle cell crisis?
Signs and symptoms can include:Anemia. Sickle cells break apart easily and die. ... Episodes of pain. Periodic episodes of extreme pain, called pain crises, are a major symptom of sickle cell anemia. ... Swelling of hands and feet. ... Frequent infections. ... Delayed growth or puberty. ... Vision problems.
What are the 4 types of sickle cell crisis?
Four major types of crises are recognised in sickle cell anaemia: aplastic, acute sequestration, hyper-haemolytic, and vaso-occlusive crises.
What is the difference between sickle cell disease and sickle cell crisis?
The reason that symptoms come and go is that the red blood cells can behave normally for much of the time - but if something makes too many of them sickle, the sickle cells cause symptoms. If there are severe and sudden symptoms due to sickling, this is called a sickle cell crisis.
What triggers sickle cell crisis?
Sickling may be triggered by conditions associated with low oxygen levels, increased blood acidity, or low blood volume. Common sickle cell crisis triggers include: sudden change in temperature, which can make the blood vessels narrow. very strenuous or excessive exercise, due to shortage of oxygen.
How long can a sickle cell patient live?
Results. Among children and adults with sickle cell anemia (homozygous for sickle hemoglobin), the median age at death was 42 years for males and 48 years for females. Among those with sickle cell-hemoglobin C disease, the median age at death was 60 years for males and 68 years for females.
Why does a sickle cell crisis last 5 7 days?
That's when you have a sickle cell crisis. The stuck cells slow or even totally block blood flow, so some parts of your body don't get the oxygen they need. That can cause intense pain that lasts anywhere from a few hours to a few weeks.
At what age does sickle cell crisis start?
People with sickle cell disease (SCD) start to have signs of the disease during the first year of life, usually around 5 months of age. Symptoms and complications of SCD are different for each person and can range from mild to severe.
Is sickle cell crisis life threatening?
Crises are a result of sickle cells pooling in the spleen. This can cause a sudden drop in hemoglobin and can be life-threatening if not treated promptly.
How do you treat sickle cell crisis?
How to Manage a Pain CrisisDrink water or other fluids when your symptoms start. Staying hydrated can help you head off the worst of an attack.Use a heating pad or take a warm bath.Try a massage, acupuncture, or relaxation techniques.Do something to take your mind off your pain.
What is the most common type of sickle cell crisis?
Sickle Cell Crises The most common is the vasoocclusive ('painful') crisis. Vasoocclusive crisis has sudden onset, usually lasts 5–6 days, and may be localized in one area of the body or generalized.
What should sickle cell patients avoid?
avoid very strenuous exercise – people with sickle cell disease should be active, but intense activities that cause you to become seriously out of breath are best avoided. avoid alcohol and smoking – alcohol can cause you to become dehydrated and smoking can trigger a serious lung condition called acute chest syndrome.
What is the most common type of sickle cell crisis?
Sickle Cell Crises The most common is the vasoocclusive ('painful') crisis. Vasoocclusive crisis has sudden onset, usually lasts 5–6 days, and may be localized in one area of the body or generalized.
At what age does sickle cell crisis start?
People with sickle cell disease (SCD) start to have signs of the disease during the first year of life, usually around 5 months of age. Symptoms and complications of SCD are different for each person and can range from mild to severe.
What does a pain crisis feel like?
Pain Crises The result is a sudden pain attack, called a pain crisis. The pain may feel sharp, stabbing, intense, or throbbing. Some people with sickle cell disease say it's worse than childbirth or the pain after surgery. You may have pain anywhere in your body and in more than one place.
How do you get rid of a sickle cell crisis?
Self-help for treating a sickle cell crisis take over-the-counter painkillers, such as paracetamol or ibuprofen (do not give aspirin to children under 16 unless a doctor has prescribed it) – if the pain is more severe, your GP may prescribe stronger painkillers. have plenty to drink.
What Is A Sickle Cell Crisis?
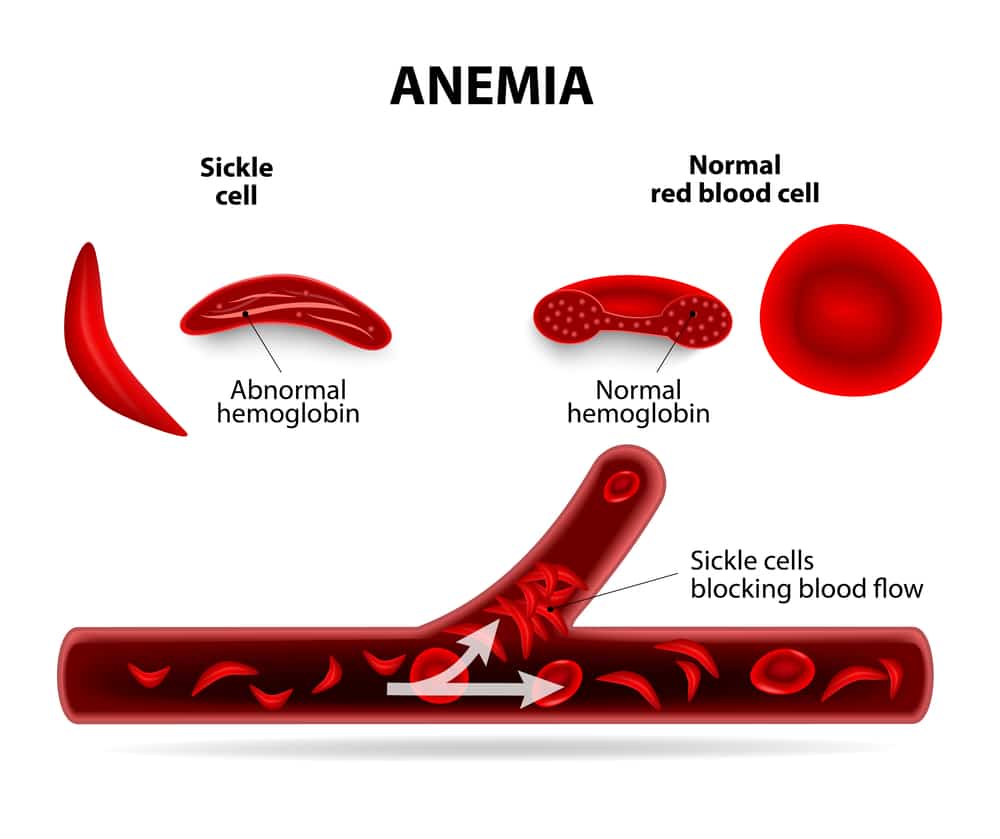
A sickle cell crisis is a painful episode that occurs in people who have sickle cell anemia. It happens when sickle-shaped red blood cells (RBCs) b...
What Are Signs and Symptoms of A Sickle Cell Crisis?
Your symptoms may change each time you have a crisis. They will depend on the area of your body where blood flow has been blocked. 1. Fever 2. Pain...
What Can Trigger A Sickle Cell Crisis?
1. Dehydration 2. Infection, such as a cold or the flu 3. Low oxygen levels from difficult exercise, flying, or high altitude 4. Getting cold or go...
How Is Pain Managed During A Sickle Cell Crisis?
1. Medicines may be given to decrease pain or to decrease sickling of your RBCs. You may also need medicine to prevent a bacterial infection or hel...
How Else Is A Sickle Cell Crisis Treated?
1. IV fluids treat dehydration and help reduce sickling of RBCs. 2. Oxygen helps increase oxygen levels in your blood and make it easier for you to...
How Can I Prevent A Sickle Cell Crisis?
1. Take vitamins and minerals as directed. Folic acid can help prevent blood vessel problems that can come with sickle cell anemia. Zinc may decrea...
Call 911 For Any of The Following
1. You have shortness of breath or chest pain. 2. You are a man and have an erection that is painful and does not go away. 3. You lose vision in on...
When Should I Seek Immediate Care?
1. You feel like you cannot cope with your pain, or you feel like hurting yourself. 2. You have behavior changes, a seizure, or faint. 3. You have...
When Should I Contact My Healthcare Provider?
1. You have any new signs or symptoms. 2. You have blood in your urine. 3. You are constipated or you have diarrhea. 4. You have changes in your vi...
How long does a sickle cell crisis last?
Sickle cell crises seem to develop in specific phases. The first phase usually lasts 1 to 2 days. During this phase, people may experience numbness, prickling, and aches in a certain area of the body. Then, a phase of increasing pain in the area occurs. Anxiety, fear, and problems with ER personnel often occur during this phase.
What causes sickle cell pain?
The pain during a sickle cell crisis is different from chronic pain, which can be caused by other complications of SCD, such as avascular necrosis of joints. 2,7.
What causes pain crises?
People with SCD experience severe acute pain because of many factors. Sickle red blood cells block blood flow because they can stick to blood vessel walls. Also, they are not flexible enough to pass through small blood vessels. This can lead to hypoxia (low oxygen levels) and ischemia (reduced blood flow), which can damage many organs. 8,9
What happens when sickle cells are blocked?
Sickle cell crises happen when blood flow and oxygen delivery are blocked by sickle cells. Pain episodes may trigger other complications of SCD, and multiple episodes can cause long-term damage. Most pain events are managed at home, but severe pain must be treated in a hospital.
Why do people with SCD go to the hospital?
The sickle cell crisis, also called acute pain crisis or vaso-occlusive crisis, is the most common reason that people with SCD go to the hospital. Episodes are sudden and unpredictable and may be triggered by different unknown risk factors. 1,2. About 5 percent of people with SCD have 3 to 10 episodes of severe pain per year.
Why do people with SCD wait so long?
However, people with SCD often see a delay in treatment for acute pain because of discrimination. People with SCD, who are mostly Black, on average wait 25 to 50 percent longer than people without SCD who have similar levels of pain. 13. Hospitalization may not be the end of the sickle cell crisis.
How to treat SCD?
Avoiding high altitudes and exposure to low oxygen. Drinking plenty of water. Getting a yearly flu shot and staying current on vaccines. Taking antibiotics to prevent infections. Taking hydroxyurea. Even though acute pain crises are the most common complication of SCD, treatment needs to be improved.
What is a sickle cell crisis?
A sickle cell crisis is a painful episode that occurs in people who have sickle cell anemia. It happens when sickle-shaped red blood cells (RBCs) block blood vessels. Blood and oxygen cannot get to your tissues, causing pain. A sickle cell crisis can also damage your tissues and cause organ failure, such liver or kidney failure. A sickle cell crisis can become life-threatening.
What are signs and symptoms of a sickle cell crisis?
Your symptoms may change each time you have a crisis. They will depend on the area of your body where blood flow has been blocked.
How is pain managed during a sickle cell crisis?
Medicines may be given to decrease sickling of your RBCs. You may also need medicine to prevent a bacterial infection or help you breathe more easily.
How to reduce sickling of RBCs?
IV fluids treat dehydration and help reduce sickling of RBCs. Oxygen helps increase oxygen levels in your blood and make it easier for you to breathe. A blood transfusion replaces blood with RBCs that are not sickle shaped. Surgery may be done to remove part of your spleen.
How to reduce pain from sickle cell?
Zinc may decrease how often you have pain. Drink liquids as directed. Dehydration can increase your risk for a sick cell crisis. Ask how much liquid to drink each day and which liquids are best for you. Balance rest and exercise. Rest during a sickle cell crisis. Over time, increase your activity to a moderate amount.
How to get rid of sickle cell?
Balance rest and exercise . Rest during a sickle cell crisis. Over time, increase your activity to a moderate amount. Exercise as directed. Avoid exercise or activities that can cause injury, such as football. Ask about the best exercise plan for you. Wash your hands frequently.
How to prevent illness?
Wash your hands frequently. Handwashing can help prevent illness. Wash your hands before you prepare or eat food, and after you use the bathroom.
How long does it take for a stuck cell to go away?
That can cause intense pain that lasts anywhere from a few hours to a few weeks. But you can take steps to lower your chances of a crisis. And even when one comes on, you may be able to care for yourself at home.
What is the best medicine for a swollen red blood cell?
Thetr are a few drugs that can help. The drug called L-glutamine oral powder ( Endari) has proven to help prevent these crises from occurring and thus preventing hospitalizations. Hydroxyurea ( Droxia , Hydrea, Silkos) and voxelotor (Oxbryta) prevent abnormal red blood cells from forming. This reduces the number of painful crises from sickling blood cells. Crizanlizumab-tmca ( Adakveo) helps stop the blood cells from sticking together and blocking small blood cells, which can not just be painful, but can damage organs.
What is sickle cell disease?
Sickle cell disease (SCD) is a term that denotes syndromes characterized by the presence of intraerythrocytic hemoglobin S (HbS), a hemoglobin tetramer composed of mutated βS-globin chains, and includes homo zygous HbS disease (HbSS) and compound heterozygous HbSC, HbS/β-thalassemia, HbSD, HbSO, and HbSE disease. The fundamental, primary lesion of SCD is the polymerization of deoxygenated HbS resulting in the deformation of RBCs into elongated, pathognomonic sickle-shaped cells. RBC sickling causes structural membrane damage leading to premature clearing of the affected cells by the reticuloendothelial system, abnormal expression of surface adhesion molecules, and impaired blood rheology. These processes are compounded by intravascular sickle cell hemolysis with release of proinflammatory heme (the tetrapyrrolic iron-binding ring in hemoglobin) and cell-free plasma hemoglobin. Hemolysis and the alterations of blood rheology stemming from a high proportion of circulating sickle cells lead to a complex inflammatory cascade affecting all components of Virchow’s triad and resulting in endothelial dysfunction, hyperviscosity, and hemostatic activation.1, 2While Virchow’s triad is traditionally invoked to explain thrombosis, a peculiar type of vascular occlusion ensues in SCD, characterized by adhesion of RBCs, leukocytes, and platelets to the endothelial wall of postcapillary venules and antegrade and retrograde vaso-occlusion.3, 4, 5, 6, 7, 8This lesion is well documented in SCD animal models, where it is responsible for organ ischemia-reperfusion injury and infarction.9Whereas RBC sickling is incessant and necessary for vaso-occlusion, vaso-occlusive crises (VOCs) are episodic, acute events that can be triggered by specific stressors in animal models and often have clear triggers in patients. Thus, patients with SCD suffer both from a chronic inflammatory vasculopathy and acute crises characterized by organ function loss. Herein, we focus on the most severe, acute manifestations of SCD that often require admission to an ICU.
What is ACS in hospital?
ACS is a lung injury syndrome that can complicate hospitalizations for acute VOC or develop de novo as a presenting admission diagnosis. It carries a high risk of respiratory failure (13% of patients requiring mechanical ventilation for a mean of 4.6 days) and a mortality of up to 9% in adults with SCD.23ACS is also associated with prolonged hospitalization (on average, 10.5 days),23, 24decreased long-term survival,25and increased risk of developing chronic lung disease.26Its presentation has a significant overlap with pneumonia because of the high prevalence of fever, dyspnea, and decreased oxygen saturation27and because infection can both trigger or complicate a prior ACS. In its classic presentation (approximately 50% of cases), ACS develops in hospitalized patients, on average on hospital day 2.5, with more than 80% of patients having experienced a prior VOC (Fig 1).23, 27aBesides the aforementioned symptoms, chest pain is particularly common in ACS and can have a pleuritic component because of the presence of extensive lung infarction. Radiographically, multilobar involvement, affecting mostly the dependent lobes in adults and adolescents, is the norm (Fig 2)28and because of its lower lobe predilection ACS is occasionally misdiagnosed as aspiration pneumonia. Patients who have new, complete lung segment consolidation on chest CT scan (approximately 60%) tend to have a more severe presentation with brisk leukocytosis and concurrent moderate to large pleural effusion and worse outcome (as indicated by the need for mechanical ventilation and extensive transfusion, and death).28Several causes of ACS have been identified, although in most cases the direct insult is unknown.23While infection is the leading offender in children,24in adults the temporal association with VOC suggests the same mechanisms can underlie both syndromes. The finding of lipid-laden alveolar macrophages by oil red O stain in up to 44% of patients with ACS who have undergone bronchoalveolar lavage has substantiated a direct causal link to VOC.29In these patients, it is hypothesized that bone marrow necrosis in the diaphysis of long bones leads to marrow fat embolization to the lungs and subsequent parenchymal ischemia and infarction. Most commonly, however, the finding of worse hemolysis in patients who develop ACS, as shown by rising lactate dehydrogenase and dropping hemoglobin levels, is indicative of the role of severe hemolysis in the causal path of ACS.30, 31This role has been definitively demonstrated in animal models of ACS where hemin, the oxidized form of heme, is both necessary and sufficient to induce ACS in sickle mice, while scavenging of free plasma hemoglobin or heme released by hemolyzed RBCs has a protective effect.32Another potential cause of ACS is hypoventilation from decreased respiratory effort, frequently resulting from either painful infarcted ribs33or opiate-induced oversedation. The evidence from a small but significant clinical trial that incentive spirometry prevents ACS lends support to the potential role of hypoventilation and atelectasis in the pathogenesis of ACS.34Finally, the finding that pulmonary thrombosis has a high prevalence in SCD35and is a common incidental finding in ACS (17% of cases)36has raised the question of whether thrombosis is a cause vs a complication of ACS. While patients with thrombosis had lower hemolysis and higher platelet counts at onset with higher peak counts,36lower platelet counts at diagnosis (< 200,000/μL) have been long recognized to herald a worse clinical course with higher risk of respiratory failure, thereby suggesting that platelet consumption from hemolysis-induced hemostatic activation is also clinically relevant. Thus, physicians should take particular note of thrombosis, although at present the evidence is not sufficient to justify obtaining a CT angiogram in all patients with ACS. Well-established diagnostic algorithms to predict pulmonary embolism in other populations include lower extremity venous Doppler ultrasound and D-dimer measurements; it is important to recognize that these algorithms are usually not helpful in SCD as pulmonary thrombosis tends to be an in situ phenomenon (without accompanying evidence of lower extremity deep vein thrombosis)36and D-dimer levels are elevated at baseline in SCD.37, 38, 39The main causes and mechanisms of ACS are outlined in Figure 1.
What is the leading cause of mortality in pediatric SCD?
Sepsis from encapsulated bacteria was the leading cause of mortality in pediatric SCD until a landmark clinical trial demonstrated the effectiveness of penicillin prophylaxis.96Universal neonatal screening programs have allowed widespread adoption of this intervention early during infancy, in combination with seven-valent conjugate pneumococcal and Haemophilus influenzaetype B vaccination, leading to a dramatic reduction of sepsis incidence97and related mortality.98The epidemiology of SCD sepsis has, therefore, changed. Most cases are now being attributed to contamination of totally implanted venous catheters (ports) routinely used for the ever-increasingly adopted chronic transfusion programs.99, 100, 101, 102In these cases, empiric antibiotic coverage for oxacillin-resistant Staphylococcus aureusshould be instituted pending the antibiogram results. In patients with recurrent sepsis and invasive infections the host susceptibility factors should be explored and, if possible, corrected. Hydroxyurea can predispose to worse sepsis outcomes by depressing the immune response, and should be discontinued in patients with active or frequent invasive infections. Iron chelation, another common therapeutic intervention in SCD, has been associated with an increased risk of sepsis from ferrophilic organisms such as Yersinia enterocolitica103, 104, 105, 106, 107and Vibrio vulnificus, but its impact on acute bacteremia and endotoxemia overall has been less clear in other patient populations.108In patients with SCD residing in or with a history of travel to a Plasmodium falciparummalaria endemic region, malarial parasitemia is associated with an increased risk for death during hospitalization (OR, 4.9),109, 110suggesting that while HbS inheritance partially protects from infection, once acute malaria develops, it portends a more ominous course.
What is the treatment for ACS?
Because of the uncertainty over the pathophysiology of ACS and the difficulty in identifying an exact trigger, treatment should target both infection and vaso-occlusion and necessarily include broad-spectrum antibiotic coverage and transfusion in all patients. Antibiotics should cover Streptococcus pneumoniae, taking into account the local antibiotic resistance patterns, and atypical microorganisms such as Chlamydia pneumoniaeand Mycoplasma pneumoniae, as these rank highly among the commonly identified pathogens (>20% of all isolates).23H1N1 pandemic influenza has also been responsible for increased morbidity and mortality in SCD, with ACS developing in 34% of patients, 17% receiving ICU care, and 10% undergoing mechanical ventilation,40thus mandating early empiric treatment with oseltamivir during seasonal outbreaks. RBC transfusions aimed at increasing oxygen delivery and reducing the percentage of HbS should be administered as simple or automated exchange transfusions (erythrocytapheresis). Automated exchange transfusions are usually required in patients with the most severe symptoms, when rapid dilution of HbS is warranted, and require placement of a double-lumen catheter and rapid mobilization of blood banking resources. They are performed by dedicated nursing staff and involve cross-matching of several units of packed red blood cells (PRBCs). An alveolar-arterial oxygen gradient > 30 mm Hg was found to be associated with worse severity score and higher need for transfusion in children,41but as a rule of thumb, patients with multilobar involvement, in respiratory distress, or who require ICU care should be aggressively treated with exchange transfusion. It is important to note, however, that simple and exchange transfusion strategies have never been directly compared in relation to ACS outcomes in a randomized trial. Potential risks of transfusion strategies in SCD are discussed in the “Hematologic Crises” section of this review. Particular care should also be afforded to patients who have compromised cardiopulmonary reserve. Approximately one-third of patients with SCD have elevated tricuspid regurgitant jet velocity (TRV) by transthoracic echocardiography at baseline, a finding that in patients with the highest elevations (> 3.0 m/s) is highly suggestive of pulmonary hypertension, and in those with more modest elevations (2.5-2.9 m/s) is linked to an increased risk for short-term mortality (rate ratio, 10.1).42In a study of patients admitted to the ICU for severe ACS the pulmonary artery systolic pressure, as estimated from the TRV, rose sharply during ACS and returned to pre-ACS values on resolution of the illness. Sudden increases in pulmonary pressures carry an increased risk of pulmonary failure and sudden death and warrant close monitoring: in the aforementioned study cor pulmonale (36% of the cases), need for invasive mechanical ventilation (16%), and hospital death (12%) occurred only in the group with TRV elevation > 3 m/s during ACS.43Furthermore, although volume expansion with crystalloids is important in SCD, it should be delivered at a cautious rate in these hemodynamically vulnerable patients, while RBC transfusions may need to be followed by diuretic therapy. Because of the established link between pulmonary thrombosis and ACS, DVT prophylaxis should be instituted routinely in patients with ACS, and clinical research is ongoing to establish whether systemic therapeutic dose anticoagulation can have a beneficial effect both in preventing thrombosis and improving the ACS disease course. Finally, although hydroxyurea has no role in the treatment of acute complications of SCD, it has been effective at preventing ACS in both children44and adults45and should be strongly recommended to any patient with SCD and a history of vaso-occlusive complications.
What causes acute chest syndrome?
These processes are responsible for acute pain and bone marrow necrosis. ACS typically occurs 2.5 days after hospitalization for a vaso-occlusive episode, and radiographically presents as new infiltrates on a chest radiograph. Common causes include fat embolization from necrotic marrow (9% of cases), pulmonary infection (30% of cases), pulmonary infarction (16%), and hypoventilation. In situ pulmonary thrombosis has been identified in 17% of patients with ACS and may also be responsible for infarction. Animal models have shown that by-products of hemolysis, such as heme, cause experimental ACS. As a result of lung injury, ventilation-perfusion mismatches and shunting ensue, with subsequent hemoglobin desaturation and hypoxemia. Tissue hypoxia in turn triggers further sickling in a vicious cycle. The chest radiographs are those of a patient with SCD who received chronic exchange transfusions (note the presence of a double-lumen “port”) on hospital day 1 (top) and day 3 (bottom). By day 3 extensive infiltrates had developed in the patient, who required endotracheal intubation for respiratory failure. PLT = platelet.
What are the triggers of acute, life-threatening VOCs?
The triggers of acute, life-threatening VOCs are protean and are discussed relative to the specific complications, but all arise from the chronic susceptibility of patients with SCD to illness. The major predisposing threats to homeostasis in SCD include immune dysfunction, chronic sterile inflammation, decreased cardiopulmonary reserve, and progressive renal deterioration. Immune dysfunction stems from functional asplenia, the result of autoinfarction of the spleen in the first years of life, and predisposes to episodes of overwhelming sepsis from encapsulated bacteria, the prime pediatric killer in SCD. Sterile inflammation has been amply documented by research showing that most proinflammatory cytokines and mediators are upregulated in SCD, leading to enhanced sensitivity to pathogenic triggers, such as respiratory allergens.10, 11, 12, 13Biventricular chamber dilatation is an early cardiac manifestation of SCD, detectable echocardiographically14and by magnetic resonance imaging15in children. It suggests wall remodeling as a compensatory mechanism to chronic anemia and progresses through adulthood. The resulting baseline elevation in cardiac output, combined with the detrimental effects of intravascular hemolysis on endothelial function, cellular proliferation, and lung parenchymal damage, eventually lead to decreased cardiopulmonary reserve, diastolic dysfunction, and pulmonary hypertension. Thus, the cardiovascular system in SCD is frequently unable to compensate for sudden increases in oxygen demand as in acute sepsis or acute anemia, predisposing patients to hemodynamic collapse in these situations.16, 17Finally, renal dysfunction is one of the first identifiable abnormalities in SCD and subtly progresses to overt kidney failure over time. A combination of glomerular hyperfiltration and tubular dysfunction, both contributing to maintain low serum creatinine levels and a relatively high estimated glomerular filtration rate in spite of progressive renal disease, lead to underdiagnosis and underestimation of renal dysfunction by medical providers until frank proteinuria and advanced kidney disease have developed.18Hemolysis, as evidenced by hemoglobinuria, has been independently associated with chronic kidney disease and can have a pathogenic role.19
Do corticosteroids help with ACS?
The routine use of systemic corticosteroids has been proposed to prevent deterioration of patients with newly diagnosed ACS and to hasten the resolution of ACS-related acute respiratory distress syndrome. Lending support to corticosteroid use is the known association between pediatric bronchial hyperreactivity and ACS58, 59, 60and the high prevalence of decreased FEV1 in adult patients with SCD.61While intravenous corticosteroids prevented deterioration and reduced the need for transfusion in ACS,62higher readmission rates among the treated patients62, 63have, unfortunately, dampened enthusiasm for the routine use of these drugs. In these cases, it has been hypothesized that rebound adhesion of demarginated leukocytes after rapid corticosteroid withdrawal may have contributed to VOC. Finally, the role of noninvasive positive-pressure ventilation in preventing and treating ACS has not been definitively investigated but holds promise.64
What is acute chest syndrome?
Acute chest syndrome is a broadly defined complication where a substance other than air is present in a section of the lung. It is a very serious condition that should be treated immediately in a hospital. In fact, ACS is the second-most common reason that people with SCD visit the hospital. It is also a leading cause of death. 1-3
What happens if you have ACS?
About 20 percent of adults with a history of ACS can experience a rapid type of ACS. This can cause quick respiratory failure and multi-organ failure, leading to kidney and liver damage. Repeated episodes of ACS can also lead to other long-term lung complications, such as pulmonary hypertension.
What percentage of people with SCD have ACS?
About 50 percent of people with SCD will have an episode of ACS. It is most common among people with sickle cell anemia (HbSS) and sickle beta zero thalassemia. 4. ACS severity increases with age because of a higher risk of arteries clogged by bone marrow and fat.
What is the best treatment for ACS?
Treatment of ACS usually involves some combination of: 10 1 Pain control with opioids 2 Increased fluid intake by mouth or IV 3 Simple or exchange blood transfusion 4 Bronchodilators, especially in adults with asthma 5 Antibiotics, especially in children 6 Oxygen supplementation 7 Steroids
Can ACS be triggered by asthma?
ACS episodes can also be triggered by infections or asthma. ACS episodes are life-threatening and must be treated immediately in a hospital. Symptoms of an ACS episode are similar to pneumonia. Prevention usually involves hydroxyurea or blood transfusions.