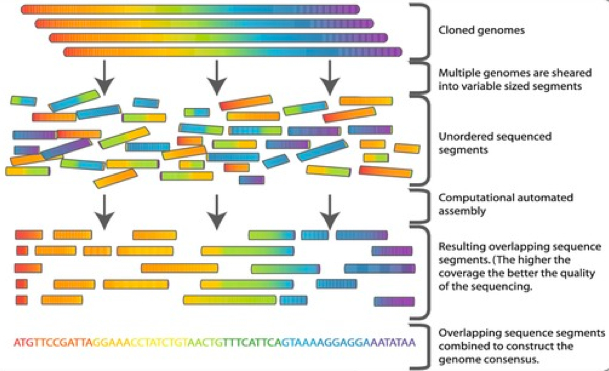
De novo sequence assembly is the process whereby we merge together individual sequence reads to form long contiguous sequences (‘contigs’) sharing the same nucleotide sequence as the original template DNA from which the sequence reads were derived.
What is de novo genome assembly?
What is de novo genome assembly? Genome assembly refers to the process of taking a large number of short DNA sequences and putting them back together to create a representation of the original chromosomes from which the DNA originated. De novo genome assemblies assume no prior knowledge of the source DNA sequence length, layout or composition.
What is de novo sequencing?
What Is De Novo Sequencing? De novo sequencing refers to sequencing a novel genome where there is no reference sequence available for alignment. Sequence reads are assembled as contigs, and the coverage quality of de novo sequence data depends on the size and continuity of the contigs (ie, the number of gaps in the data).
What is Dedede novo assembly of sequence reads?
De novo assembly of sequence reads generated by classical Sanger capillary sequencing is a mature field of research. Unfortunately, the existing sequence assembly programs were not effective for short sequence reads generated by Illumina and SOLiD platforms.
What is genome assembly and how is it done?
Genome assembly refers to the process of taking a large number of short DNA sequences and putting them back together to create a representation of the original chromosomes from which the DNA originated [1]. De novo genome assemblies assume no prior knowledge of the source DNA sequence length, layout or composition.
How do you assemble a genome de novo?
The protocol in a nutshell:Obtain sequence read file(s) from sequencing machine(s).Look at the reads - get an understanding of what you've got and what the quality is like.Raw data cleanup/quality trimming if necessary.Choose an appropriate assembly parameter set.Assemble the data into contigs/scaffolds.More items...
What is a de novo approach?
De novo methods utilize computational approaches to deduce the sequence or partial sequence of peptides directly from the experimental MS/MS spectra. The concepts behind a number of de novo sequencing methods are discussed.
What is sequence assembly in bioinformatics?
In bioinformatics, sequence assembly refers to aligning and merging fragments from a longer DNA sequence in order to reconstruct the original sequence.
Which assembly algorithm is best for de novo assembly?
Hybrid assembling algorithm refers to the mixing various assembling algorithms. It is used to reduce the number of contigs and errors produced by other algorithms. There are many de novo assemblers available online which have been developed by applying one of these five assembling algorithms.
What is the definition de novo?
Primary tabs. De novo is a Latin term that means "anew," "from the beginning," or "afresh." When a court hears a case “de novo,” it is deciding the issues without reference to any legal conclusion or assumption made by the previous court to hear the case.
What is the meaning of Novo?
Latin. adverb. anew; afresh; again; from the beginning.
Why sequence assembly is important?
DNA sequence assembly is a process that involves aligning and merging fragments of a DNA sequence to reconstruct the original structure of the DNA. This is an essential step of the genome analysis process because the entire genome cannot be interpreted in one step with current sequencing technology.
How is sequence assembly done?
Sequence assembly can be done using one of three approaches: (1) greedy, (2) overlap-layout-consensus (OLC) and Hamiltonian path, and (3) de Bruijn graph and Eulerian pathd.
Which type of read is good choice for de novo genome assembly?
There are two basic approaches in algorithms for short-read assem- blers: overlap graphs and de Bruijn graphs. These approaches are described below. High quality de novo sequence assembly using Illumina Genome Analyzer reads is possible today using publicly available short-read assemblers.
What is a contig in sequencing?
A contig (as related to genomic studies; derived from the word “contiguous”) is a set of DNA segments or sequences that overlap in a way that provides a contiguous representation of a genomic region.
What is the use of genome assembly?
In bioinformatics, genome assembly represents the process of putting a large number of short DNA sequences back together to recreate the original chromosomes from which the DNA originated.
What is a good N50 value?
Contiguity is often measured as contig N50, which is the length cutoff for the longest contigs that contain 50% of the total genome length. In this era of long-read genome assemblies, a contig N50 over 1 Mb is generally considered good.
What is the purpose of resequencing?
With targeted resequencing, a subset of genes or regions of the genome are isolated and sequenced. Targeted approaches using next-generation sequencing (NGS) allow researchers to focus time, expenses, and data analysis on specific areas of interest.
What is a contig in sequencing?
A contig (as related to genomic studies; derived from the word “contiguous”) is a set of DNA segments or sequences that overlap in a way that provides a contiguous representation of a genomic region.
What is the difference between de novo and salvage pathway?
The salvage pathway uses free bases via a reaction with phosphoribosyl pyrophosphate (PRPP) and generation of nucleotides. De novo pathways synthesize pyrimidines and purine nucleotides from amino acids, carbon dioxide, folate derivatives, and PRPP.
What is the difference between ab initio and de novo?
Ab initio structure prediction classically refers to structure prediction using nothing more than first-principles (i.e. physics). De Novo is a more general term that refers to the greater category of methods that do not use templates from homologous PDB structures.
What is de novo sequencing?
De novo sequencing refers to sequencing a novel genome where there is no reference sequence available for alignment. Sequence reads are assembled as contigs, and the coverage quality of de novo sequence data depends on the size and continuity of the contigs (ie, the number of gaps in the data).
Who uses de novo sequencing to understand the evolution of the Kiwi bird?
Dr. Diana Le Duc uses de novo sequencing to understand the evolution of the kiwi bird.
What is Illumina sequencing?
Illumina is providing sequencing for a UK-wide study led by Genomics England, designed to compare the genomes of severely and mildly ill COVID-19 patients. This research may help uncover genetic factors associated with susceptibility.
When sequencing a genome for the first time, a combined approach can yield higher-quality assemblies.?
When sequencing a genome for the first time, a combined approach can yield higher-quality assemblies. For example, combining short-insert, paired-end and long-insert, mate pair sequences is one option to maximize coverage. The short reads, sequenced at higher depths, can fill in gaps not covered by the long inserts.
What is the purpose of sequencing sweet potato viruses?
Scientists at the Agricultural Research Council in South Africa use sequencing to identify known and novel sweet potato viruses, with the goal of enhancing food security. Read Article.
What is the goal of a sequence assembler?
The goal of a sequence assembler is to produce long contiguous pieces of sequence (contigs) from these reads. The contigs are sometimes then ordered and oriented in relation to one another to form scaffolds. The distances between pairs of a set of paired end reads is useful information for this purpose.
What is genome assembly?
Genome assembly refers to the process of taking a large number of short DNA sequences and putting them back together to create a representation of the original chromosomes from which the DNA originated [1]. De novo genome assemblies assume no prior knowledge of the source DNA sequence length, layout or composition.
Why is DNA sequence important?
Because of the importance of DNA to living things , knowledge of a DNA sequence may be useful in practically any biological research. For example, in medicine it can be used to identify, diagnose and potentially develop treatments for genetic diseases. Similarly, research into pathogens may lead to treatments for contagious diseases [2].
Why is genome assembly so difficult?
Genome assembly is a very difficult computational problem, made more difficult because many genomes contain large numbers of identical sequences, known as repeats. These repeats can be thousands of nucleotides long, and some occur in thousands of different locations, especially in the large genomes of plants and animals. [1]
Do parameters have an effect on assembly?
These parameters can and do have a large effect on the outcome of any assembly. Assemblies can be produced which have less gaps, less or no mis-assemblies, less errors by tweaking the input parameters. Therefore, knowledge of the parameters and their effects is essential to getting good assemblies.
What is a de novo assembler?
The Geneious de novo assembler parses your input data and will select the appropriate Sensitivity: setting to use. In most cases you will not need to adjust the Sensitivity setting. The assembler also estimates and reports the amount of memory expected to be required to perform the assembly. The assembler will warn you if it believes you will not have enough RAM to assemble your selected data set.
How many files are written in assembly?
Upon completion of assembly three new files will be written, an assembly report, an assembly file, and a consensus sequence generated from the assembly.
What assembler is used in Exercise 2?
In Exercise 2 we will assemble the trimmed paired reads using the Geneious de novo assembler.
How to install Geneious Prime tutorial?
Complete the tutorial yourself with included sequence data. Download the tutorial then install by dragging and dropping the zip file into Geneious Prime. Do not unzip the tutorial.
What is the average insert size of Illumina MiSeq?
These data comprise paired Illumina MiSeq reads with a raw read length of 149 bp and an expected average insert size of 350 bp. The sequences are derived from the the genome of Escherichia coli str. K-12 substr. MG1655 (Full genome available at NC_000913 ).
Why is preprocessing important for NGS?
Proper preprocessing of your NGS reads will improve assembly accuracy and also will usually significantly reduce the computation and time required to complete assembly.
How many sections are there in the settings window?
The settings window is divided into 5 sections, Data , Method , Trim , Results and, via the More Options button, an Advanced Settings section. For an overview of the various settings see section 10.3 of the online manual.
What is de novo genome assembly?
De novo (from new) genome assembly refers to the process of reconstructing an organism’s genome from smaller sequenced fragments. This is not a trivial task, and can involve multiple types of data and analysis methods/tools. A good analogy of this task is the example below Sequence Assembly Wiki.
Is there a best approach to genome analysis?
There is no “best” approach, or a golden standard, when assembling a genome. Ideally, you want to use multiple tools/parameters and explore what best works in each individual case, and this can be a lengthy and sometimes complex analysis.
What is de novo sequencing?
De novo sequence assembly is the process whereby we merge together individual sequence reads to form long contiguous sequences (‘contigs’) sharing the same nucleotide sequence as the original template DNA from which the sequence reads were derived . Assembly of sequence reads generated by classical Sanger capillary sequencing is a mature field of research [ 17 ]. Two main types of algorithm are commonly used: (i) the overlap-layout-consensus (OLC) approach and (ii) algorithms based on a de Bruijn graph. These have been well reviewed previously [ 15–20] and have been implemented in very effective genome-assembly software packages including Arachne [ 24 ], AMOS [ 25 ], Atlas [ 26 ], Celera Assembler [ 27 ], CAP3 [ 28 ], Euler [ 29 ], PCAP [ 30 ], Phrap [ 31 ], RePS [ 32] and Phusion [ 33 ]. Unfortunately, these genome sequence assembly programs are not well suited to short sequence reads generated by Illumina and SOLiD platforms, for several reasons. First, the numbers of reads produced by Illumina and SOLiD systems are around three to four orders of magnitude greater per run per instrument per day than that from capillary sequencers. OLC methods require overlaps to be scored between all possible pairs of reads, which is computationally costly, scaling with the square of the number of reads. Second, the shorter sequences mean any base-calling errors have a much greater potential impact. Third, unique overlaps between pairs of reads are much less likely, given the short read-lengths. Finally, it is often not possible to resolve repetitive sequences where the length of the repeated unit is longer than the sequence read. Given these challenges, most recent de novo genome sequencing projects have opted for Roche’s 454 GS-FLX sequencing technology (rather than the Illumina or the SOLiD platforms). These decisions were probably influenced by its significantly lower cost per nucleotide (compared to capillary sequencing) yet longer read-length (compared to Illumina and SOLiD) and the availability of Roche’s proprietary ‘Newbler’ assembler, which is optimized for GS-FLX data. Another approach has been to perform ‘hybrid’ assemblies of mixtures of both long and short reads in an attempt to gain the respective benefits of both (e.g. [ 34–38 ]). However, given the even lower per-nucleotide costs of Illumina and SOLiD, some researchers, especially those with access to a local instrument, have been tempted to use these short-read platforms for de novo genome sequencing. This has led to the generation of several draft genome sequences ( Table 1) [ 39–48] based exclusively on short sequence Illumina sequence reads, recently culminating in the assembly of the 2.25 Gb genome of the giant panda from Illumina sequence reads with an average length of just 52 nucleotides [ 40, 49 ]. Of the currently published genome assemblies built exclusively from short reads, most are no more than a few megabases in length and they still contain on the order of hundreds of gaps. The assembly of the panda genome from short reads, although it is a momentous achievement, contains significantly more gaps than previous mammalian genome assemblies based on longer reads and questions have been raised about its completeness and accuracy [ 49 ].
What is sequence assembly?
The sequence assembly problem is essentially one of constructing a DNA sequence superstring that explains the observed set of sequence reads. This superstring might represent the chromosome, bacterial artificial chromosome, or other template DNA that was subjected to sequencing. If the data were completely error-free, then we would expect every sequence read to be contained within the superstring. So, we might be tempted to formulate sequence assembly as finding a superstring that contains all sequence-read strings as substrings. In real biological sequence data, the problem is more complicated. Sequencing error rates may be as high as 1–4% per nucleotide implying that many of the sequence reads contain mismatches with respect to the solution superstring. Furthermore, there will inevitably be multiple solutions; that is, we could propose many possible superstrings that satisfy the criterion of containing all the observed sequence reads. So which superstring is the best one? In a spirit of parsimony, we might choose the shortest superstring, but this would likely not be the biologically correct one. The reason is that real genomic DNA sequences tend to contain large numbers of perfectly and/or imperfectly repeated sequences, which would be erroneously collapsed in the shortest superstring. Some of the major differences between the popular assembly programs are to be found in their strategies for dealing with repeats. For example, whilst the Celera assembler [ 27] masks-out repeat sequences, EULER proactively utilizes repeats to determine contig order. A further complication is that sequence reads can originate from the reverse complement as well as from the forward orientation of the template DNA sequence.
How to compare quality of an assembly?
There are two dimensions of quality: contiguity and accuracy . Contiguity refers to the lengths of the contigs and/or the scaffolds. In practice, the set of contigs comprising an assembly will not be of uniform length, so the measure of contiguity is essentially a description of a distribution of lengths. Therefore, useful metrics might include the mean, median, minimum and maximum lengths. However, arguably the most useful summary statistic is the N50 length. N50 is calculated by first ordering all contigs (or scaffolds) by length and then summing their lengths (starting with the longest) until the sum exceeds 50% of the total length of all contigs. Alternatively, where the length of the target genome is known, then N50 calculation is sometimes based on 50% of the genome length rather than 50% of the sum of contig lengths [ 108 ]. The N50 contig (or scaffold) number is the number of contigs (or scaffolds) of at least N50 in length.
What software packages can be used to assemble Illumina short sequence reads?
These software packages are able to perform de novo assembly of Illumina short sequence reads with the exception of SHORTY, which is designed to assemble ABI SOLiD colour-space data. Velvet and SOPRA can assemble sequence-space and colour-space data. a Curtain is a pipeline, based on Velvet, for hierarchical assembly of short sequence reads in order to overcome memory usage limitations. b Oases is specifically designed for assembling transcribed sequences.
What is the new generation of sequencing?
A new generation of sequencing technologies is revolutionizing molecular biology. Illumina’s Solexa and Applied Biosystems’ SOLiD generate gigabases of nucleotide sequence per week. However, a perceived limitation of these ultra-high-throughput technologies is their short read-lengths. De novo assembly of sequence reads generated by classical Sanger capillary sequencing is a mature field of research. Unfortunately, the existing sequence assembly programs were not effective for short sequence reads generated by Illumina and SOLiD platforms. Early studies suggested that, in principle, sequence reads as short as 20–30 nucleotides could be used to generate useful assemblies of both prokaryotic and eukaryotic genome sequences, albeit containing many gaps. The early feasibility studies and proofs of principle inspired several bioinformatics research groups to implement new algorithms as freely available software tools specifically aimed at assembling reads of 30–50 nucleotides in length. This has led to the generation of several draft genome sequences based exclusively on short sequence Illumina sequence reads, recently culminating in the assembly of the 2.25-Gb genome of the giant panda from Illumina sequence reads with an average length of just 52 nucleotides. As well as reviewing recent developments in the field, we discuss some practical aspects such as data filtering and submission of assembly data to public repositories.
How many nucleotides are in a sequence read?
Early studies suggested that, in principle, sequence reads as short as 20–30 nucleotides could be used to generate useful assemblies of both prokaryotic and eukaryotic genome sequences, albeit containing many gaps.
What are the recent innovations in bioinformatics and in vitro library preparation?
Recent innovations in bioinformatics and in vitro library preparation promise to bring short read sequence assembly up to a comparable level with longer-read technologies with respect to assembly quality.
What is genome assembly?
De novo genome assembly is a strategy for genome assembly, representing the genome assembly of a novel genome from scratch without the aid of reference genomic data. De novo genome assemblies assume no prior knowledge of the source DNA sequence length, layout or composition.
Is Illumina sequencing a common approach?
We will take the Illumina genome assembly as an example to introduce the workflow of genome assembly with NGS data, since Illumina sequencing is one of the most common approaches for genomics studies.
