Researchers participating in a clinical trial must report all adverse events to the drug regulatory authority of the respective country where the drug or device is to be registered [e.g. Food and Drug Administration (FDA) if it is US]. Serious AEs must be reported immediately; minor AEs are 'bundled' by the sponsor and submitted later.
Do patients have to report adverse events?
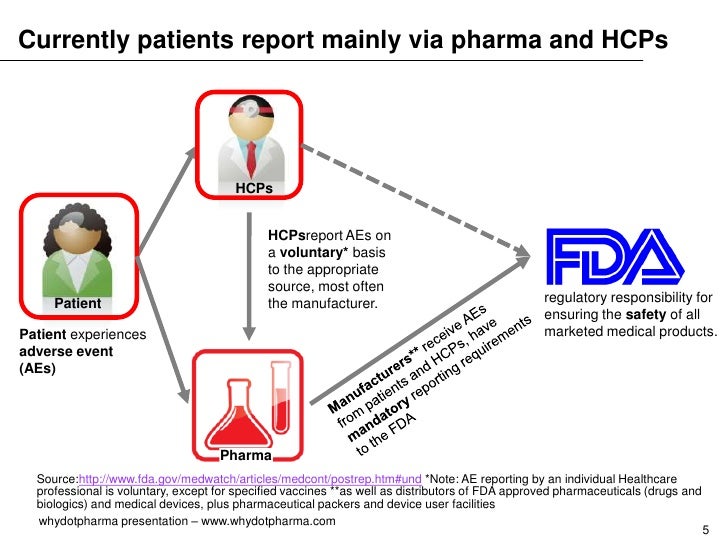
Patients have no legal requirement to report adverse events, but healthcare providers and medical manufacturers are subject to mandatory reporting requirements. Medical product reports are submitted through the MedWatch Voluntary Reporting Form , and clinical vaccine reports go through the Vaccine Adverse Event Reporting System (VAERS).
What is the FDA adverse event reporting system?
The FDA Adverse Event Reporting System (FAERS) is a database that contains adverse event reports, medication error reports and product quality complaints resulting in adverse events that were submitted to FDA. The database is designed to support the FDA's post-marketing safety surveillance program for drug and therapeutic biologic products.
Are all adverse events (AE) reported?
Nevertheless, we encounter investigators and also sponsors classifying AE as not related to the study treatment without unbinding in function of pre-established rules in the protocol. That misinterpretation in the definition of AE reporting brings up the issue of how to ensure that all AE are reported.
How do you determine if an adverse event was due to?
First, there is no certainty that the reported event (adverse event or medication error) was due to the product. FDA does not require that a causal relationship between a product and event be proven, and reports do not always contain enough detail to properly evaluate an event.

Who can report an adverse event AE in clinical trial?
The medical management of the AE/ADR rests on the investigator. According to the DCR-6th Amdmt,[3] the investigator should report all SAEs to the drug regulatory body of India (DCGI), sponsor of the trial, and the concerned EC that approved the trial protocol within 24 h of occurrence of the SAE.
Who is responsible for reporting an adverse drug reaction?
Physicians' professional commitment to advance scientific knowledge and make relevant information available to patients, colleagues, and the public carries with it the responsibility to report suspected adverse events resulting from the use of a drug or medical device.
Who is required to report adverse events to FDA?
FDA has determined that dietary supplement manufacturers, packers, and distributors must report serious adverse events associated with their products using either the paper MedWatch form, Form FDA 3500A or the FDA Safety Reporting Portal.
How do you report an adverse drug event?
Use one of the methods below to submit voluntary adverse event reports to the FDA:Report Online.Consumer Reporting Form FDA 3500B. ... Call FDA at 1-800-FDA-1088 to report by telephone.Reporting Form FDA 3500 commonly used by health professionals.
How are adverse drug reactions reported FDA?
To do so: complete and submit the report online at www.fda.gov/medwatch/report.htm; or. download and complete the form, then submit it via fax at 1-800-FDA-0178.
Who can report pharmacovigilance?
9. Indian Pharmacopoeia Commission, Pharmacovigilance Programme of India Who can report? All healthcare professionals (Clinicians, Dentist, Pharmacist, Nurses, Physician, Physiotherapist etc) • All non- healthcare professionals including consumers/ patients etc can report ADRs.
When should you report an adverse event?
Fatal or life-threatening serious unexpected suspected adverse reactions (SUSARs) reports: The sponsor (or sponsor-investigator) must notify the FDA of any SUSARs to the study drug as soon as possible but no later than 7 calendar days after the initial receipt of the information.
When should ADR be reported?
Serious, unexpected reactions (ADRs) that are not fatal or life-threatening must be filed as soon as possible but no later than 15 calendar days after first knowledge by the sponsor that the case meets the minimum criteria for expedited reporting.
What is considered serious medical event?
Important medical events that may not result in death, be life-threatening, or require hospitalization may be considered serious when, based upon appropriate medical judgment, they may jeopardize the patient and may require medical or surgical intervention to prevent one of the outcomes listed above.
How to contact VAERS?
If you need further assistance with reporting to VAERS, please email [email protected] call 1-800-822-7967.
Who sponsors VAERS?
VAERS is co-sponsored by the Centers for Disease Control and Prevention (CDC), and the Food and Drug Administration (FDA), agencies of the U.S. Department of Health and Human Services (HHS).
What is an adverse event?
An adverse event occurs when a patient encounters any undesirable experience associated with the use of a drug or medical device. They range from unconcerning to life-threatening, and they can even include events like changing the color of a person's urine. If you learned about allergies to specific antibiotics as a child, ...
What Happens to Reports?
Once made, regulators at the Center for Drug Evaluation and Research (CDER) or the Center for Biologics Evaluation and Research (CBER) analyze all reports to determine which may legitimately be caused by the drug or device in question. Sometimes they find that the negative symptom occurred naturally or stemmed from some other cause. When the adverse effect seems to be caused by the medical device or drug, the FDA may follow-up with one of these options to protect public health and safety:
What is FDA safety?
According the the United States Food and Drug Administration (FDA), all drug and device safety information is thoroughly vetted before a product gains approval. Unfortunately, even well-designed clinical studies can miss problems, meaning that some issues emerge only after a product has been released for public use. As such, it is important for all consumers to understand what constitutes an adverse event, how they are monitored, and how everyone can help protect public health by reporting the negative effects of drugs and medical devices.
What are the factors that influence the symptoms of a medical product?
Especially when the general population uses medical products, a number of variables can influence the symptoms experienced after using them, such as lifestyle, diet, other medical conditions, and environmental factors. Some patients may face unintended side effects simply as a result of underlying conditions. ...
Do you have to report adverse events to a medical provider?
Patients have no legal requirement to report adverse events, but healthcare providers and medical manufacturers are subject to mandatory reporting requirements. Medical product reports are submitted through the MedWatch Voluntary Reporting Form, and clinical vaccine reports go through the Vaccine Adverse Event Reporting System (VAERS).
Is it important to notify the FDA of side effects?
If your symptom is on the label, it's still important to notify the FDA. Sometimes side effect frequency is underestimated in clinical trials. This kind of report can help both regulators and manufacturers update labeling information to more accurately represent side effect probability.
Can you report adverse events on Essure?
If women had known they could voluntarily report their negative experiences with the Essure implantable birth control device from the outset, it may have been pulled from the market much earlier and prevented countless injuries. Remember, adverse event reporting helps keep us all safe, and it's really easy to do.
How to report adverse events to the FDA?
Use one of the methods below to submit voluntary adverse event reports to the FDA: 1 Report Online 2 Consumer Reporting Form FDA 3500B. Follow the instructions on the form to either fax or mail it in for submission. For help filling out the form, see MedWatch Learn. 3 Call FDA at 1-800-FDA-1088 to report by telephone 4 Reporting Form FDA 3500 commonly used by health professionals. View Instructions for Form FDA 3500
What to do if you have a serious reaction to a medical product?
If you think you or someone in your family has experienced a serious reaction to a medical product, you are encouraged to take the reporting form to your doctor. Your health care provider can provide clinical information based on your medical record that can help FDA evaluate your report.
How to contact the FDA about a product?
If you need information or if you have questions or comments about a medical product, please call the FDA's toll-free information line, 1-888-INFO-FDA (1-888-463-6332) Press 2 to report into MedWatch or for instructions.
How to report FDA 3500B?
Consumer Reporting Form FDA 3500B. Follow the instructions on the form to either fax or mail it in for submission. For help filling out the form, see MedWatch Learn. Call FDA at 1-800-FDA-1088 to report by telephone. Reporting Form FDA 3500 commonly used by health professionals. View Instructions for Form FDA 3500.
What are healthcare providers encouraged to report?
Healthcare providers are encouraged to report: Any adverse event that occurs after the administration of a vaccine licensed in the United States, whether or not it is clear that a vaccine caused the adverse event. Vaccine administration errors.
What is a vaer?
The Vaccine Adverse Event Reporting System (VAERS) is an early warning system that helps CDC and the Food and Drug Administration (FDA) monitor health problems that may occur following vaccination. VAERS is a passive surveillance system, and relies on people sending in reports of their experiences. As the frontline system for vaccine safety ...
Where to report AE?
While an AE may be reported to the manufacturer, to FDA (e.g., via MedWatch), or to the registry itself (and then from the registry to the manufacturer), it is strongly encouraged that the protocol describe the procedures that should be followed, and that the sites be trained in these procedures as well as in their general obligations and the relevant public health considerations. A separate safety reporting plan that fully identifies the responsible parties and describes the operational considerations may also be considered to ensure that potentially reportable information is evaluated in an appropriate timeframe, and, for manufacturer-sponsored registries, in accordance with any applicable standard operating procedures. This type of plan also should describe how deviations or systemic failures in detection and reporting processes will be identified, addressed, and considered for corrective action.
What is AE reporting?
All AE reporting begins with a suspicion by the physician (or responsible person who obtains or receives information) that a patient exposed to a medicinal product has experienced some AE and that the event has a reasonable possibility of being causally related to the product being used ; this is referred to as the “becoming aware” principle. Some registries also collect and record AEs reported directly by the patients or their caregivers. It is important to develop a plan for detecting, processing, and reporting AEs for any registry that has direct patient contact. If the registry receives sponsorship in whole or part from a regulated industry (for drugs or devices), the sponsor has mandated reporting requirements, including stringent timelines. AE reporting requirements for registry sponsors are discussed later in this chapter.
How long does it take to report a AE?
For registries, the 15-calendar-day notification applies if the regulated industry believes there is a reasonable possibility that the unexpected SAE was causally related to product exposure. Best practices for international reporting are that all “affiliates” of a sponsor report serious, unexpected, and possibly related events to the sponsor in a timely fashion, ideally within 2 calendar days; this allows the sponsor, in turn, to complete notification to the responsible regulatory authority within a total of 15 calendar days. Events that do not meet the requirements of expedited reporting (such as nonserious events or serious events considered expected or not related) may require submission through inclusion in an appropriate safety update, such as the New Drug Application or Biologic Licensing Application Annual Report, Periodic Report, or Periodic Safety Update Report, as applicable.4In many cases, sponsors are also required to provide registry safety updates to the health authority. Thus, sponsors may coordinate registry safety updates (i.e., determining the date for creating the dataset—the data cutoff date) with the timing of the New Drug Application Annual Report, Periodic Report, Periodic Safety Update Report, or other agreed-upon periodic reporting format. Devices, however, have different reporting requirements (see http://www.fda.gov/MedicalDevices/Safety/ReportaProblem/default.htm). In any event, sponsors should discuss safety reporting requirements for their specific registries with the applicable health authorities (such as FDA and European Medicines Agency) before finalizing their registry protocol.
What is AE in medical records?
Registries that collect information on specific drugs and medical devices need to anticipate the need for adverse event (AE) detection, processing, and reporting. This chapter addresses the identification, processing, and reporting of AEs detected in situations in which a registry has contact with individual patients. This document is not a formal regulatory or legal document; therefore, any information or suggestions presented herein do not supersede, replace, or otherwise interpret Federal guidance documents that touch on these subjects. Registry sponsors are encouraged to discuss plans for AE collection and processing with local health authorities when planning a registry.
What is the minimum dataset required to report an AE?
The minimum dataset required to consider information as a reportable AE is indeed minimal, namely (1) an identifiable patient, (2) an identifiable reporter, (3) product exposure, and (4) an event . However, in addition to direct data collection, AEs can be detected through retrospective analysis of a population database, where direct patient or health care provider contact does not occur. Patient interactions include clinical interactions and data collection by phone, Internet, or other means; perusal of electronic medical records or insurance claims data would not be considered direct patient interaction. Reporting is rarely required for individual AEs observed in aggregate population data, since there is no direct patient interaction where an association might be suggested or inferred. Nevertheless, if aggregate or epidemiologic analyses suggest that an AE is associated with exposure to a drug or medical product, it is desirable that the minimum dataset information be forwarded to the manufacturer of the product, who will determine any need for, and timing of, reporting of study results to the relevant regulatory authorities.
How to report an AE to FDA?
Once suspicion has been aroused that an unexpected serious event has a reasonable possibility of being causally related to a drug, the AE should be reported to FDA through MedWatch, to the company that manufactures the product, or to the registry coordinating center. (See Chapter 11.) A system should be developed such that all appropriate events are captured and duplicate reporting is avoided to the extent possible. Generally, AE reports are submitted directly by the site or by the registry to the manufacturer, since they are often most efficient at evaluating, processing, and reporting for regulatory purposes within the required time periods. Alternatively, sites could be instructed to report AEs directly to FDA according to their normal practices for marketed products; however, this often means that the companies are not notified of the AE and are not able to follow up or evaluate the event in the context of their safety database. In fact, companies are not necessarily notified by FDA if an AE report comes directly to FDA, since only certain reports are shared with industry, and reporters have an option to request that the information not be shared directly with the company.14When sites report AEs directly to FDA, this process can also lead to inadvertent duplication of information for events recorded both by the registry and the company.
What is an adverse drug experience?
The U.S. Food and Drug Administration (FDA) defines an adverse drug experience as any AE associated with the use of a drug in humans, whether or not considered drug related,4while the International Conference on Harmonisation (ICH) guideline ICH E2A similarly defines an AE as an untoward medical occurrence in a patient administered a pharmaceutical product, whether or not the occurrence is related to or considered to have a causal relationship with the treatment.5
How can I report an adverse event or medication error to FDA?
The MedWatch website provides information about voluntary and mandatory reporting.
Who sends reports to FAERS?
Healthcare professionals, consumers, and manufacturers submit reports to FAERS. FDA receives voluntary reports directly from healthcare professionals (such as physicians, pharmacists, nurses and others) and consumers (such as patients, family members, lawyers and others). Healthcare professionals and consumers may also report to the products’ manufacturers. If a manufacturer receives a report from a healthcare professional or consumer, it is required to send the report to FDA as specified by regulations.
How do I find or confirm my report is in FAERS?
To confirm that your report is in FAERS, please send a Freedom of Information (FOI) request to FDA.
Is the FAERS public dashboard accessible on an Android™ or iPhone®?
Yes, but the user interface layout may not be very user friendly. FDA will continue to work on the dashboard to make the user interface Android and iPhone friendly.
Where else can I find safety information?
Potential Signals of Serious Risks/New Safety Information Identified from the FDA Adverse Event Reporting System (FAERS): quarterly reports on potential serious side effects identified by FAERS.
What is post-marketing drug and biologic safety evaluation?
Post-marketing Drug and Biologic Safety Evaluations: provides summary information about ongoing and completed post-marketing safety evaluations of adverse experience reports made to FDA for New Drug Applications (NDAs) and Biologic License Applications (BLAs) approved since September 27, 2007.
What is duplicate reporting?
Duplicate reporting occurs when the same report is submitted by the consumer and the sponsor. The information in FAERS evolves daily and the number of individual cases may increase or decrease.
What is an adverse event?
An adverse event (AE), as defined by Good Clinical Practice, is any unfavorable and unintended sign (including an abnormal laboratory finding), symptom, or disease having been absent at baseline, or, if present at baseline, appears to worsen AND is temporally associated with medical treatment or procedure, REGARDLESS of the attribution (i.e. , relationship of event to medical treatment or procedure).
What is the importance of monitoring adverse events?
Monitoring of adverse events (AEs) is critical to the patient’s safety (i.e., human subjects protection) and data integrity. This module will provide an overview of AEs, including assessment, documentation, recording, and reporting.
What does a sponsor report?
Sponsor must also report expeditiously any findings from clinical, epidemiological, or pooled analysis of multiple studies or any findings from animal or in vitro testing that suggest a significant risk in humans exposed to the drug
What is an adverse reaction?
Adverse reaction, the nature or severity of which is not consistent with the applicable product information (e.g., Investigator's Brochure for an unapproved investigational product or package insert/summary of product characteristics for an approved product).
What does "unexpected" mean in a drug report?
Unexpected,” also refers to AEs or SARs mentioned in the investigator brochure as occurring with a class of drugs or as anticipated from the pharmac ological properties of the drug, but are not specifically mentioned as occurring with the particular drug under investigation
What is considered serious medical event?
Important medical events that may not result in death, be life-threatening, or require hospitalization may be considered serious when, based upon appropriate medical judgment, they may jeopardize the patient or subject and may require medical or surgical intervention to prevent one of the outcomes listed in this definition. Examples include:
How long does it take to notify FDA of a SAR?
Sponsor must notify FDA of any unexpected fatal or life-threatening SAR as soon as possible but in no case later than 7 calendar days after the sponsor's initial receipt of the information
What is an adverse event?
Adverse event (AE) is an absolute term in function to Good Clinical Practices, defined as “… any untoward medical occurrence in a patient or clinical investigation subject ( when) administered a pharmaceutical product and that does not necessarily have a causal relationship with this treatment”.
Who is responsible for assigning significance to all lab abnormalities and reporting them in the case report form?
The investigator is responsible to assign significance to all lab abnormalities and report them in the Case Report Form.
What happens if a lab abnormality meets exclusion?
If the lab abnormality meets exclusion, the monitor should procure patient withdrawal. Lab results are the bulk of safety data, and proper review, classification and reporting should be ensured to avoid overlooking safety signals.
Why do monitors not challenge lab abnormality classification?
In most of the cases, monitors do not challenge a lab abnormality classification, as well as the investigator only review the lab results in function of the safety of the patient, but not classify abnormalities because considers that all was already electronically classified and submitted by the lab automatically. They might occasionally override an abnormal classification to allow patient eligibility in a clinical trial when the value is not significant.
What is the role of a clinical trial monitor?
Unreported or underreported adverse events are a major problem during regulatory inspections. The monitor has the responsibility to capture any issue with reporting before the inspector does, and ensure compliance.
What is an AE?
Adverse event (AE)is an absolute term in function to Good Clinical Practices, defined as “… any untoward medical occurrence in a patient or clinical investigation subject (when) administered a pharmaceutical product and that does not necessarily have a causal relationship with this treatment”. This is different from the definition of adverse drug reaction(ADR) that is “…all noxious and unintended responses to a medicinal product related to any dose (…)means that a causal relationship between a medicinal product and an adverse event is at least a reasonable possibility…”
Why is it important for a monitor to verify documentation behind the investigators classification before they concur?
The reason behind it is to allow further reassessment when the clinical trial/study report is written and when submission of safety data is performed.