What are CYP450 drug interactions?
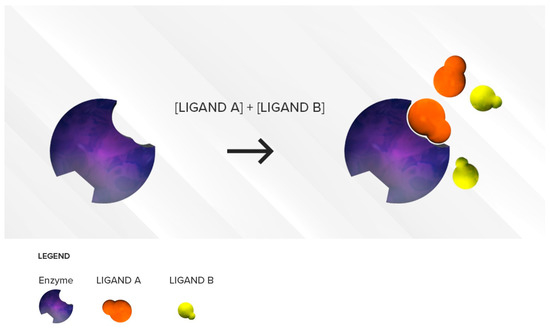
The cytochrome P450 (CYP450) enzymes are essential to produce numerous agents, including cholesterol and steroids. They are also necessary for the detoxification of foreign chemicals and the metabolism of drugs. Drugs that cause CYP450 drug interactions are referred to as either inhibitors or inducers.
What is the role of cytochrome P450 in drug metabolism?
Abstract. Cytochrome P450 is a family of isozymes responsible for the biotransformation of several drugs. Drug metabolism via the cytochrome P450 system has emerged as an important determinant in the occurrence of several drug interactions that can result in drug toxicities, reduced pharmacological effect, and adverse drug reactions.
Is CYP450 inhibition reversible?
In some cases, CYP450 inhibition is irreversible. The formation of a stable complex, between a drug and the metabolizing enzyme, is one mechanism that can result in irreversible inhibition. 1 The inhibitor can be a drug or one of its metabolites.
Where can I find information about a drug's CYP450 metabolism?
Information regarding a drug's CYP450 metabolism and its potential for inhibition or induction can be found on the drug label and accessed through the U.S. Food and Drug Administration (FDA) or manufacturer's websites.
See more

What happens when you inhibit CYP450?
Cytochrome P450 enzymes can be inhibited or induced by drugs, resulting in clinically significant drug-drug interactions that can cause unanticipated adverse reactions or therapeutic failures. Interactions with warfarin, antidepressants, antiepileptic drugs, and statins often involve the cytochrome P450 enzymes.
What does CYP450 enzyme do?
Cytochrome P450 monooxygenases catalyze the oxidation and metabolism of a large number of xenobiotics and endogenous compounds. CYP450 enzymes evolved as the primary defense against xenobiotics and in this process are also responsible for the bioactivation of drugs and toxicants to more reactive intermediates.
What happens when CYP3A4 is inhibited?
1-4 The inhibition of CYP3A4 can result in the accumulation of parent drug concentrations that can put the patient at increased risk for side effects and possible toxicity.
What does CYP450 inducer mean?
Cytochrome P450 enzymes are essential to metabolise many medications. Cytochrome P450 (CYP450) enzymes can be inhibited or induced by some drugs, resulting in significant drug interactions that can cause unanticipated adverse reactions or therapeutic failures.
What is the role of cytochrome P450 in detoxification?
Insect cytochrome P450 monooxygenases (P450s) are well known to be involved in metabolic detoxification of xenobiotics, such as phytochemicals, insecticides and environmental pollutants. Enhanced metabolic detoxification is closely associated with the constitutive overexpression and induction of P450s.
How do you remember CYP450 inhibitors?
0:512:39Cytochrome P450 Enzyme Inhibitors - Easy Mnemonic - YouTubeYouTubeStart of suggested clipEnd of suggested clipWe can easily remember this mnemonic Vic's. Face all over GQ stuffs ladies in the track. Where VMoreWe can easily remember this mnemonic Vic's. Face all over GQ stuffs ladies in the track. Where V from VIX is for vamp red eyes for isoniazid sis for cimetidine case for ketoconazole S is for
What drugs are broken down by P450?
Clinical example of P450-based interactionsTerfenadine. Terfenadine is the first non-sedating H1-antihistamine drug. ... Cimetidine. Cimetidine inhibits antihistamine H2-receptor binding and is used in the treatment of gastric ulcers. ... Grapefruit juice. ... Omeprazole. ... Erythromycin. ... Cyclosporin. ... Rifampicin.
What is the role of CYP3A4?
CYP3A4 is a major cytochrome P450. It catalyses a broad range of substrates including xenobiotics such as clinically used drugs and endogenous compounds bile acids. Its function to detoxify bile acids could be used for treating cholestasis, which is a condition characterised by accumulation of bile acids.
What is the difference between enzyme inducer and inhibitor?
Enzyme inhibitor is a molecule that decreases the activity of an enzyme by binding with the active site of the enzyme while enzyme inducer is a molecule that increases the metabolic activity of an enzyme either by binding to it or by increasing the gene expression.
Is grapefruit a CYP450 inhibitor or inducer?
Grapefruit juice inhibits the CYP3A4 enzyme of the cytochrome P450 system in the intestinal mucosa, increasing the bioavailability of drugs with a high first pass metabolism. Therapeutic doses of the affected drugs may produce serious adverse reactions.
What does it mean if a drug is an inducer?
Drugs that cause CYP450 drug interactions are referred to as either inhibitors or inducers. An inducing agent can increase the rate of another drug's metabolism by as much as two- to threefold that develops over a period of a week.
Which drug is an inducer of CYP450 enzymes?
Cimetidine, a typical CYP450 enzyme inducer, can increase both plasma concentration and elimination half-life, but the clinical significance of this is unclear.
What are cytochromes and what is their purpose?
cytochrome, any of a group of hemoprotein cell components that, by readily undergoing reduction and oxidation (gain and loss of electrons) with the aid of enzymes, serve a vital function in the transfer of energy within cells. Hemoproteins are proteins linked to a nonprotein, iron-bearing component.
What drugs are metabolized by CYP450?
Among the drugs metabolized are sedatives such as midazolam, triazolam and diazepam, the antidepressives amitriptyline and imipramine, the anti-arryhthmics amiodarone, quinidine, propafenone and disopyramide, the antihistamines terfenadine, astemizole and loratidine, calcium channel antagonists such as diltiazem and ...
What does CYP3A4 do in the human body?
CYP3A4 is a major cytochrome P450. It catalyses a broad range of substrates including xenobiotics such as clinically used drugs and endogenous compounds bile acids. Its function to detoxify bile acids could be used for treating cholestasis, which is a condition characterised by accumulation of bile acids.
Where are CYP450 enzymes found?
Within humans, CYP enzymes are mainly found within the endoplasmic reticulum and mitochondria of liver cells. However, they are also found in many other cells of the body. These membrane-bound proteins are involved in the metabolism of many harmful substrates, such as toxins.
What is the role of CYP450 isoforms in drug interactions?
The ability of a single CYP450 isoform to metabolize multiple substrates is responsible for several drug interactions associated with reversible competitive inhibition. Competitive inhibition occurs when two substrates compete for the same active site—such as the prosthetic heme iron or substrate-binding region—of CYP450s. The competition is a function of the respective affinities of the two substrates for the binding site and their concentrations in the proximity of the enzyme. First, the most clinically relevant situation will be discussed.
What is cytochrome P450 inhibitory?
Inhibition of cytochrome P450 (CYP450) enzymes is the most common mechanism leading to drug–drug interactions [4] . CYP450 inhibition can be categorized as reversible (including competitive and non-competitive inhibition) or irreversible (or quasi-irreversible), such as mechanism-based inhibition. Each type of interaction involves a distinct clinical management strategy. This is why a comprehensive understanding of mechanisms of CYP450-mediated metabolism inhibition is needed to prevent or mitigate these harmful drug interactions. This review will focus on the CYP450 enzymatic system with a special look at one specific type of CYP450 inhibition, namely mechanism-based inhibition; clinical cases involving mechanism-based inhibitors will be discussed in this context.
What are the consequences of polypharmacy?
One common consequence of polypharmacy is the increased emergence of adverse drug events, mainly from drug–drug interactions. The majority of currently available drugs are metabolized by CYP450 enzymes. Interactions due to shared CYP450-mediated metabolic pathways for two or more drugs are frequent, especially through reversible or irreversible CYP450 inhibition. The magnitude of these interactions depends on several factors, including varying affinity and concentration of substrates, time delay between the administration of the drugs, and mechanisms of CYP450 inhibition. Various types of CYP450 inhibition (competitive, non-competitive, mechanism-based) have been observed clinically, and interactions of these types require a distinct clinical management strategy. This review focuses on mechanism-based inhibition, which occurs when a substrate forms a reactive intermediate, creating a stable enzyme–intermediate complex that irreversibly reduces enzyme activity. This type of inhibition can cause interactions with drugs such as omeprazole, paroxetine, macrolide antibiotics, or mirabegron. A good understanding of mechanism-based inhibition and proper clinical management is needed by clinicians when such drugs are prescribed. It is important to recognize mechanism-based inhibition since it cannot be prevented by separating the time of administration of the interacting drugs. Here, we provide a comprehensive overview of the different types of mechanism-based inhibition, along with illustrative examples of how mechanism-based inhibition might affect prescribing and clinical behaviors.
What is reversible inhibition?
Reversible inhibition is a result of rapid association and dissociation between the substrate drugs and the enzyme and can be categorized as competitive or non-competitive. A third category, uncompetitive inhibitor, also considered as a reversible inhibition type, is a very rare phenomenon and will not be considered in this current review; this type of inhibitor binds only the enzyme–substrate complex, leading to a dead-end complex.
How does a decrease in the CLintof affect the drug?
For an active drug, the decrease in the CLintof one of its metabolic pathways can lead to a decrease in the total clearance of the drug (capacity for eliminating the drug) and can result in increased plasma concentrations, potentially precipitating adverse effects. However, for prodrugs, this interaction can instead result in a decrease in the formation of the active metabolite, reducing drug efficacy. The magnitude of changes observed in the overall disposition of the victim drug (increase in its plasma levels) will be a function of the relative contribution of the inhibited metabolic pathway to the clearance of this drug. For example, if 15% of a drug is excreted unchanged in urine—35% by enzyme 1 and 50% by enzyme 2—a 50% decrease in the total CLof the victim drug is expected if enzyme 2 is inhibited:
Is theophylline a substrate of CYP1A2?
In an example illustrating this scenario, theophylline (weak CYP1A2 substrate) is largely metabolized by CYP1A2 by binding to its active site. (Figure 3) If that active site is occupied by a stronger substrate like duloxetine (moderate CYP1A2 affinity substrate), breakdown of theophylline will be reduced (↓CLint), leading to increased plasma levels of theophylline and possibly side effects (e.g., headache, nausea, vomiting). To minimize competitive inhibition, two competing substrates should be administered with as much time apart as possible.
Is CYP450 mediated metabolism reversible?
Mechanisms of CYP450 inhibition can be categorized as reversible (including competitive or non-competitive) or irreversible/quasi-irreversible ( mechanism-based inhibition).
How to determine the contribution of a specific CYP?
The contribution of a specific CYP can be given directly by the percentage of inhibition of the NCEs metabolism. The most common CYP specific inhibitors are given in Table 4.
What drugs can prolong QTC?
TCAs are known to prolong the QTc interval, so care should be taken with the concomitant use of other agents associated with QT prolongation, potentially proarrhythmic medications, sodium channel blockers, cardiac glycosides, and noradrenergic drugs, such as cocaine and amphetamines.
How is 20-HETE biosynthesis regulated?
Endothelial cell and vascular 20-HETE biosynthesis can be regulated by genetically manipulating CYP4A and CYP4F expression ( Wu & Schwartzman, 2011 ). CYP4A and CYP4F enzymes have been knocked down or overexpressed in cell culture systems ( Roman, 2002; Wu & Schwartzman, 2011 ). Expression of CYP4A2 in vivo in rat endothelial cells has been accomplished by adenovirus intravenous injection ( Wu et al., 2013 ). Antisense nucleotide CYP4A1/CYP4A2 inhibition in spontaneously hypertensive rats (SHRs) resulted in decreased 20-HETE levels ( Sodhi et al., 2010 ). The CYP4A enzyme has been genetically manipulated in the Dahl salt-sensitive (SS) rat by introgression of CYP4A alleles for the normotensive Brown-Norway rat into the Dahl SS genetic background ( Lukaszewicz & Lombard, 2013 ). This manipulation resulted in a decrease in 20-HETE production in the Dahl SS rat ( Lukaszewicz & Lombard, 2013 ). Cyp4a genes have also been manipulated in mice. Cyp4a14 gene deficiency in mice resulted in increased Cyp4a12 (a) expression and increased 20-HETE levels ( Holla et al., 2001 ). On the other hand, Cyp4a10 gene deficiency in mice did not change Cyp4a12 (a) expression or 20-HETE levels ( Capdevila, Pidkovka, Mei, et al., 2014 ). Interestingly, Cyp4a10 −/− mice had decreased Cyp2c44 epoxygenase expression ( Capdevila et al., 2014 ). These findings highlight the complex regulation of CYP hydroxylase and epoxygenase enzymes and provide evidence to interactions between these enzymatic pathways. The overall impact of interactions between hydroxylase and epoxygenase pathways on vascular and endothelial study is not completely understood.
What is the role of C24 in oxidation?
The hypothesis that the main role of the C24-oxidation pathway is attenuation of the 1,25 (OH) 2D biological signal inside target cells was tested in vitro using cytochrome P450 inhibitors. Blocking P450 activity by treatment of cells with the antifungal imidazole derivative, ketoconazole, inhibits catabolism and results in 1,25 (OH) 2 D accumulation and extended hormone action [38]. This hypothesis was also tested and confirmed in vivo by engineering Cyp24a1 -deficient mice to examine the role of the CYP24A1 enzyme in vitamin D homeostasis [39].
How long does phenobarbital last?
With regard to pharmacokinetics, the half-life is 10 to 20 hours; up to 95% of the drug is protein bound, a lower percentage at higher doses; as a partial cytochrome P450 inhibitor, it causes elevation particularly of phenobarbital and lamotrigine levels.
Which tricyclic drugs have the least interaction with TCAs?
The degree of drug interactions with TCAs is largely dependent on the specific tricyclic agent used and its relative inhibitory potential of hepatic CYP 450 oxidative enzymes. Desipramine and nortriptyline have the least drug interactions because they are weak CYP 450 2D6 inhibitors and are unlikely to be involved in clinically relevant interactions unless the serum levels are high—for example, following overdose or in “slow hydroxylators.” Amitriptyline, imipramine, clomipramine, dothiepin, and doxepin, all tertiary amines, are more potent CYP 450 inhibitors and therefore are more problematic in terms of drug interactions. 81
Is cresol a toxic isomer?
The comparative toxic potential of cresol isomers (o-, m-, and p -met hylphenol) was demonstrated in rat liver slices. The toxicity induced by p-methylphenol could be prevented by pretreatment of the slices with the thiol precursor N -acetylcysteine, and was enhanced with the glutathione-depleting agent diethyl maleate. 80
What is the role of cytochrome P450?
Cytochrome P450 (CYP) is a hemeprotein that plays a key role in the metabolism of drugs and other xenobiotics (Estabrook, 2003). Understanding the CYP system is essential for advanced practitioners (APs), as the consequences of drug-drug interactions can be profound. In this article, we will describe the CYP system, its potential for drug interactions, and the genetic polymorphisms that can exist in hematology/oncology patients.
Why is CYP important?
It is the root of many drug interactions due to inhibition, induction, and competition for common enzymatic pathways by different drugs. Genetic variability of CYP is also a significant source of unpredictable drug effects. Awareness and understanding of drugs involved in common CYP pathways will add to the AP’s knowledge base to foresee and prevent potential drug interactions and untoward effects.
How does genetic polymorphism affect drug therapy?
Genetic polymorphisms can have a significant impact on drug therapy and should be taken into consideration in clinical practice, especially when unexpected outcomes arise. For example, intermediate and poor metabolizers are at increased risk for toxicity and adverse effects due to drug accumulation. These patients demonstrate hypersensitivity or low tolerance to particular drugs and subsequently may require reduced doses or avoidance of the drug altogether. Acute dystonic reactions due to metoclopramide administration have been documented in patients with homozygous CYP2D6 variant alleles (van der Padt, van Schaik, & Sonneveld, 2006). Similarly, deaths have been reported in patients receiving methadone and fluoxetine, in separate cases. Autopsies revealed abnormally high drug concentrations that were attributed to the presence of CYP2B6 and CYP2D6 variant alleles, respectively (Bunten, Liang, Pounder, Seneviratne, & Osselton, 2010; Sallee, DeVane, & Ferrell, 2000). Conversely, prodrugs, defined as inactive parent drugs that require enzymatic conversion to the active metabolite, may exhibit low drug efficacy in poor metabolizers. These patients may need higher doses of drugs to produce the same response as extensive metabolizers.
What is the metabolite of codeine?
Codeine is metabolized to the active metabolite morphine via CYP2D6 and generally provides mild analgesic and cough suppressant effects. However, when codeine is administered to patients carrying CYP2D6 gene duplication, approximately 50% more morphine is produced (Kirchheiner et al., 2007). As a consequence of such amplification of drug effects, devastating outcomes have occurred. For example, a baby suffered from fatal morphine toxicity when her mother was prescribed codeine while breastfeeding. Genotyping later showed that the mother carried an extra copy of the wild-type CYP2D6 gene (Madadi et al., 2007). Likewise, tramadol, another commonly prescribed analgesic drug, is metabolized via CYP2D6 to a more active agent. Respiratory depression and increased adverse effects have been reported in ultrarapid metabolizers (Stamer, Stuber, Muders, & Mushoff, 2008). Alternatively, effects of certain drugs may be diminished or short-acting due to rapid metabolism and deactivation in these patients.
What are the potential pitfalls of CYP?
Drug interactions are not the only potential pitfalls related to CYP. Genetic mutations or polymorphisms (genetic variants) of CYP are known to exist among patients. Depending on the phenotype encoded by these genes, the metabolism of certain drugs may be variable. Each person’s ability to metabolize drugs is determined by the pairing of individual alleles he or she has inherited from his or her parents. Each allele may be categorized as a wild-type (functional) or variant (defective) allele. Wild-type alleles are considered "normal" and occur predominantly in the general population, whereas variant alleles may confer diminished or no activity. People who carry two wild-type alleles will generally have "normal" rates of metabolism (extensive metabolizers), whereas a person who carries two variant (defective) alleles will inherently have little to no enzyme activity (poor metabolizers). Those who inherited one of each allele will have decreased enzymatic activity (intermediate metabolizers). In certain cases, when gene duplication or amplification results in more than two gene copies of wild-type alleles, enzyme activity will be greater than normal (ultrarapid metabolizers; Johansson & Ingelman-Sundberg, 2011).
How are cytochrome P450 pathways classified?
Cytochrome P450 pathways are classified by similar gene sequences; they are assigned a family number (e.g., CYP1, CYP2) and a subfamily letter (e.g., CYP1A, CYP2D) and are then differentiated by a number for the isoform or individual enzyme (e.g., CYP1A1, CYP2D6). Drugs that share a common pathway have the potential for drug-drug interactions (Nelson, 2009). The classification of CYP proteins will be the APs first hint of the potential for drug interactions. Not all drugs have CYP activity. However, drugs with CYP activity may be inhibitors, inducers, or substrates for a specific CYP enzymatic pathway, thus altering the metabolism of concurrently administered agents. Drugs that inhibit an enzymatic pathway of CYP may cause increased concentrations of other drugs metabolized by the same pathway, resulting in drug toxicity. Likewise, drugs that induce an enzymatic pathway of CYP may reduce concentrations of drugs metabolized by the same pathway, leading to subtherapeutic drug levels or treatment failure.
When was CYP first discovered?
Klingenberg first discovered CYP in 1954 during his research on steroid hormone metabolism, when he extracted a novel protein from hepatocytes (Klingenberg, 1958). It was almost a decade later that the function and significance of CYP were determined. In 1963, Estabrook, Cooper, and Rosenthal described the role of CYP as a catalyst in steroid hormone synthesis and drug metabolism. Cooper and colleagues later confirmed CYP to be a key enzyme involved in drug and steroid hydroxylation reactions (Cooper, Levin, Narasimhulu, Rosenthal, & Estabrook, 1965). Numerous CYP proteins have since been discovered and found to be widespread throughout the body, demonstrating significant involvement in chemical activation, deactivation, and carcinogenesis (Estabrook, 2003).
How long does it take for a CYP450 inhibitor to onset?
Because many drugs have relatively short half-lives (<12 hours), the onset and offset will occur over 2 to 4 days of starting or discontinuing the precipitant drug, respectively. In some cases, CYP450 inhibition is irreversible.
How long does CYP450 last?
CYP450 enzyme half-life in humans is about 36 hours ; thus, 3 to 5 days may be required for enzyme function to return to baseline following the discontinuation of an irreversible inhibitor. Cytochrome enzyme inhibition can occur by several mechanisms. The result is an increase in the concentration of the object drug.
What happens when the concentration of a precipitant drug falls?
As the concentration of the precipitant drug falls, the object drug is again able to bind to the enzyme, and the interaction abates. The timing of the onset and offset of the inhibition is dependent on the half-life of the precipitant drug. Because many drugs have relatively short half-lives ...
What is reversible inhibition?
Reversible inhibition is perhaps the most common mechanism of drug-drug interactions. Reversible inhibition implies that the effect of the precipitant drug on the enzyme metabolizing the object drug is the result of mutually exclusive competition between the precipitant drug and the object drug for binding to the enzyme.
When does competitive inhibition fail?
Competitive inhibition fails to occur when 2 drugs that are substrates for the same enzyme are coadministered when there is sufficient enzyme to accommodate both drugs. Competitive inhibition is observed when the plasma concentration (and/or binding affinity) of one drug is much greater than that of the second drug.
Do irreversible inhibitors require more time to onset?
Irreversible inhibitors may require more time for drug interaction onset and offset than competitive inhibitors, and often result in a greater change in object drug clearance. Drs. Horn and Hansten are both professors of pharmacy at the University of Washington School of Pharmacy.
Can two drugs have the same metabolic pathway?
Although any 2 drugs that have the same metabolic (enzyme) pathway can display competitive inhibition, the interaction only becomes an issue when the concentration and the binding affinity of one drug is high enough to block access to the enzyme by the other drug. Competitive inhibition fails to occur when 2 drugs that are substrates for ...
What is the role of cytochrome P450 in drug metabolism?
Predominantly operating within hepatocytes, their principal function is to metabolize hosts of xenobiotics and clearance of potentially toxic compounds. While paramount in many aspects of human biology and essential life processes, its most significant feature rests in the field of pharmacokinetic research and drug metabolism; this is the primary area with which drug developers and researchers concern themselves as different drugs can potentially affect the activity of CYP enzymes, or conversely, be affected by CYP activity, leading to unforeseen clinical outcomes. Understanding mechanisms underlying a drug’s action on these enzymes is essential for patients to receive adequate therapy, especially in conjunction with other CYP metabolized drugs. Other features of CYP enzymes rest in their ability to synthesize and break down hormones, fat-soluble vitamin metabolism, fatty acid regulation, and clearance of various toxic endogenous and exogenous compounds.[1]
What is the role of CYP in catalyzing reactions?
As monooxygenases, CYP's primary role is the addition of an oxygen atom to the substrate. In general, the mechanism CYP exhibits in catalyzing reactions depends on a few simple steps.
Why are CYP enzymes important?
The various effects of medications and other compounds on CYP enzymes are key in drug development to determine their safety and efficacy in the general public. Certain drugs are known inhibitors and inducers of specific CYP enzymes and require careful monitoring in patients taking multiple agents metabolized by the same subfamily. Two isozymes, CYP3A4 and CYP2D6, make up the bulk of drug metabolism, and drugs that interact with these enzymes should, therefore, merit closer evaluation and monitoring. [2]
What are the CYP enzymes?
Within cells, CYP enzymes closely associate with the endoplasmic reticulum and inner mitochondrial membrane activity. The enzymes surrounding the endoplasmic reticulum further express their action to metabolize external substances such as drugs, whereas the mitochondrial enzymes focus on internal substances such as steroid hormone metabolism and fatty acid regulation. Activation of these enzymes is prompted by transcription, as evidenced by varying degrees of expression in correspondence to mRNA levels. [7]
What is the role of genes in CYP?
Additionally, genes play a role in an individual’s natural composition of CYP enzymes and determine their rate of metabolism. Looking into the future of medical advancements regarding pharmacogenomics, genetic polymorphisms of CYP alleles could take precedent in determining the proper therapy and drug dose for individual patients. With this information, we would better be able to predict drug response in patients.
Where is cytochrome P450 found?
While present in most body tissues, CYP enzymes predominantly occupy the liver, intestines, and kidneys, with their highest concentration in the liver. Of the total 57 isozymes discovered to date, 6 of these are responsible for 90% of drug metabolism. These six isozymes include CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4.[2] Various drugs work on different isozymes, and determining which isozymes are affected is critical in drug development. [3]
What is the purpose of drug metabolism?
The purpose of drug metabolism is to safely administer the active constituent(s) accordingly and transform an exogenous compound into a more hydrophilic substance. Once it is water-soluble, drugs and their metabolic byproducts travel to the kidneys, where they are then filtered and excreted from the body. [8][9]Drug metabolism can classify under three different phases in which the CYP system is responsible for phase I. Reactions that occur during this phase include oxidation, reduction, and hydrolysis - oxidation being the chief mode of metabolism.
What is the CYP450 system?
The CYP450 enzymes are essential for the production of numerous agents including cholesterol and steroids.
Why are CYP450 enzymes important?
Additionally, these enzymes are necessary for the detoxification of foreign chemicals and the metabolism of drugs. CYP450 enzymes are so named because they are bound to membranes within a cell (cyto) and contain a heme pigment (chrome and P) that absorbs light at a wavelength of 450 nm when exposed to carbon monoxide.
How many CYP450 enzymes are there?
There are more than 50 CYP450 enzymes, but the CYP1A2, CYP2C19, CYP2D6, CYP1A2, CYP3A4, and CYP3A5 enzymes are responsible for metabolizing 45% of drug metabolism. The CYP2D6 (20–30%), the CYP2C9 (10%), and the CYP2E1 and CYP1A2 (5%) complete this enzyme system.
What medications interact with grapefruit juice?
In the case of grapefruit juice, there are numerous medications known to interact with grapefruit juice including statins, antiarrhythmic agents, immunosuppressive agents, and calcium channel blockers.
Does tamoxifen affect CYP2D6?
The researchers concluded that CYP2D6 metabolism is an "independent predictor of breast cancer outcome in post-menopausal women receiving tamoxifen for early breast cancer. Determination of CYP2D6 genotype may be of value in selecting adjuvant hormonal therapy and it appears CYP2D6 inhibitors should be avoided in tamoxifen-treated women." Do oncology patients come to us with only their cancer and its treatment? No, they come with multifaceted dimensions and co-morbid conditions such as hypertension, dyslipidemia, depression, seizure disorders, etc. For example, several antidepressants (paroxetine [Paxil] and fluoxetine [Prozac]) are inhibitors of metabolism when given with drugs metabolized through the CYP2D6 enzyme, such as haloperidol (Haldol), metoprolol (Lopressor), and hydrocodone. Thus, the therapeutic response can be accentuated. Medications that inhibit the CYP3A4 enzyme, such as amiodarone and antifungals, can affect the therapeutic response of fentanyl, alprazolam (Xanax), and numerous statins; as a result, the effect of these drugs can be enhanced leading to potential toxic levels.
What drugs inhibit CYP3A4?
Medications that inhibit the CYP3A4 enzyme, such as amiodarone and antifungals, can affect the therapeutic response of fentanyl, alprazolam (Xanax), and numerous statins; as a result, the effect of these drugs can be enhanced leading to potential toxic levels.
Can you take CYP450 inhibitors with tobacco?
At times, these CYP450 inducers and inhibitors are commonly ingested items such as grapefruit juice and tobacco. In the case of grapefruit juice, there are numerous medications known to interact with grapefruit juice including statins, antiarrhythmic agents, immunosuppressive agents, and calcium channel blockers.
Pharmacogenetics
One out of every 15 white or black persons may have an exaggerated response to standard doses of beta blockers (e.g., metoprolol [Lopressor]), or no response to the analgesic tramadol (Ultram). This is because drug metabolism via CYP450 enzymes exhibits genetic variability (polymorphism) that influences a patient's response to a particular drug. 3
Adverse Drug Effects
Standard drug doses may cause adverse effects related to elevated drug serum levels if a person is a poor metabolizer or has a CYP450 enzyme inhibitor added to therapy. 5, 29 Adverse effects are more likely to occur if a drug has a narrow safety range or is dependent on only one enzyme for metabolism.
What is the role of P450 in drug metabolism?
Drug metabolism via the cytochrome P450 system has emerged as an important determinant in the occurrence of several drug-drug interactions. A greater degree of interaction predictability has been achieved through the identification of P450 isozymes and some of the drugs that share them. Six different P450 isozymes—CYP1A2, CYP2C19, CYP2C9, CYP2D6, CYP2E1, and CYP3A4—that play important roles in drug metabolism have been identified (1, 2). Of these 6 isozymes, shared metabolism by the CYP3A4 isozyme has resulted in several clinically significant drug-drug interactions. More information about the effects of certain drugs on enzyme-mediated biotransformation has led to identification of enzyme inducers and inhibitors, providing even greater insight into the nature of the interactions.
What is the role of cytochrome P450 in biotransformation?
Cytochrome P450 represents a family of isozymes responsible for biotransformation of many drugs via oxidation.
What is the cytochrome P450?
Abstract. Cytochrome P450 is a family of isozymes responsible for the biotransformation of several drugs. Drug metabolism via the cytochrome P450 system has emerged as an important determinant in the occurrence of several drug interactions that can result in drug toxicities, reduced pharmacological effect, and adverse drug reactions.
What are the effects of CYP3A4 isozyme on the elderly?
In the elderly, decreases in hepatic blood flow, enzyme activity, and liver mass result in reduced metabolic activity. CYP3A4 ISOZYME INTERACTIONS. Studies on the CYP3A4 isozyme and drug-drug/drug-food interactions are becoming an integral part of drug research.
What are the substrates of CYP3A4?
For CYP3A4-metabolized drugs that require periodic monitoring of serum levels, the interaction of another CYP3A4- metabolized drug can be controlled by dosage adjustments to maintain appropriate levels of the monitored drug. Cyclosporine (CYA), tacrolimus, and carbamazepine are all substrates of CYP3A4. Coadministration of cyclosporine with a CYP3A4 inhibitor decreases an individual's CYA dosage requirement. Drinking grapefruit juice may be an inexpensive way to reduce cyclosporine dosages, but the unpredictable nature of the inhibition of CYA metabolism has not vindicated this practice. Ketoconazole and diltiazem, purer entities of CYP3A4 inhibitors, have been used successfully in this respect. Patients unable to obtain therapeutic CYA levels with orally administered cyclosporine due to inadequate absorption can been placed on either of these agents to achieve this goal.
Where is the cytochrome P450 located?
The enzymes are heme-containing membrane proteins, which are located in the smooth endoplasmic reticulum of several tissues. Although a majority of the isozymes are located in the liver, extrahepatic metabolism also occurs in the kidneys, ...
What is the problem with prescribing drugs that share the CYP3A4 pathway?
When the serum levels of these drugs reach a toxic state, the toxicity can manifest itself with serious medical consequences.
What is CYP3A4?
CYP3A4 is one of the cytochrome P450 monooxygenases (CYPs), which are enzymes that eliminate most of the drugs and toxins from our body [ 1 ].
What does inhibiting the activation of some medications that are administered as pro-drugs do?
Inhibiting the activation of some medications that are administered as pro-drugs and thus decreasing the actual dose of the active form of the drug that reaches the blood; this lowers the efficacy of the drug [ 13 ].
What enzyme is responsible for clearing drugs?
Different supplements, food components, and drugs can change CYP3A4 activity and, as a result, interfere with drug metabolism. Find out more about its function, gene variants, and factors that decrease/increase CYP3A4 activity.
Where is CYP found?
This enzyme is mainly found in the liver (∼40% of the total liver CYP content) but also in the small intestine, prostate, breast, colon, and brain [ 7, 6, 4, 3 ].
Does CYP3A4*22 cause hot flashes?
CYP3A4*22 carriers were less likely to have severe hot flashes as side effects on tamoxifen therapy (132 patients) [ 8 ].
Does curcumin increase or decrease CYP3A4?
Curcumin has a paradoxical effect, since it both increases and decreases the activity of CYP3A4 [ 61, 62, 63 ].
