Protein secondary structure prediction (PSSP) is a fundamental task in protein science and computational biology, and it can be used to understand protein 3-dimensional (3-D) structures, further, to learn their biological functions. In the past decade, a large number of methods have been proposed for PSSP.
What does protein structure, secondary mean?
The secondary structure of a protein is due to the folding of the polypeptide chain into different folds due to hydrogen bonding and Vander Waal forces. Whereas the tertiary structure of proteins is defined as the arrangement of secondary structure content in 3-dimensional space.
What is the secondary structure of protein?
The term secondary structure refers to the interaction of the hydrogen bond donor and acceptor residues of the repeating peptide unit. The two most important secondary structure of proteins, the alpha helix, and the beta sheet were predicted by the American chemist Linus Pauling in the early 1950s.
Do all proteins have secondary and tertiary structure?
All proteins have primary, secondary and tertiary structures but quaternary structures only arise when a protein is made up of two or more polypeptide chains. The folding of proteins is also driven and reinforced by the formation of many bonds between different parts of the chain.
What is the difference between primary and secondary protein?
Proteins structures are made by condensation of amino acids forming peptide bonds. The sequence of amino acids in a protein is called its primary structure. The secondary structure is determined by the dihedral angles of the peptide bonds, the tertiary structure by the folding of proteins chains in space.

What is protein structure prediction used for?
Protein structure prediction can be used to determine the three-dimensional shape of a protein from its amino acid sequence1. This problem is of fundamental importance as the structure of a protein largely determines its function2; however, protein structures can be difficult to determine experimentally.
What does secondary protein structure mean?
Secondary structure refers to regular, local structure of the protein backbone, stabilised by intramolecular and sometimes intermolecular hydrogen bonding of amide groups. There are two common types of secondary structure (Figure 11). The most prevalent is the alpha helix.
Why do we need to predict the secondary structure of a protein?
Protein secondary structure prediction provides a significant first step toward tertiary structure prediction, as well as offering information about protein activity, relationships, and functions. Protein secondary structure refers to the local conformation proteins' polypeptide backbone.
Which method is used for secondary structure prediction?
The GOR method of protein secondary structure prediction is described. The original method was published by Garnier, Osguthorpe, and Robson in 1978 and was one of the first successful methods to predict protein secondary structure from amino acid sequence.
What is the difference between primary and secondary protein structure?
The primary structure is comprised of a linear chain of amino acids. The secondary structure contains regions of amino acid chains that are stabilized by hydrogen bonds from the polypeptide backbone. These hydrogen bonds create alpha-helix and beta-pleated sheets of the secondary structure.
What are three different types of secondary protein structure?
There are three common secondary structures in proteins, namely alpha helices, beta sheets, and turns.
What are the methods for protein structure prediction?
There are three major theoretical methods for predicting the structure of proteins: comparative modelling, fold recognition, and ab initio prediction.
What is meant by structure prediction?
Definition. Structure prediction is the prediction of the three-dimensional structure of materials, such as crystals, proteins or small molecules.
What is secondary structure analysis?
Secondary Structure (SS) SS refers to highly regular local sub-structures. Two main types of SS, the alpha helix and the beta strand, were suggested in 1951 by Linus Pauling and coworkers. These SS are defined by patterns of hydrogen bonds between the main-chain peptide groups.
What is secondary structure determined by?
The secondary structure of proteins is determined by the pattern of hydrogen bonding. A large number of server and tools are used to predict the secondary structure analysis.
How are secondary structures formed?
The secondary structure arises from the hydrogen bonds formed between atoms of the polypeptide backbone. The hydrogen bonds form between the partially negative oxygen atom and the partially positive nitrogen atom.
How protein structure is determined?
To determine the three-dimensional structure of a protein at atomic resolution, large proteins have to be crystallized and studied by x-ray diffraction. The structure of small proteins in solution can be determined by nuclear magnetic resonance analysis.
What is meant by secondary and tertiary structure of proteins?
A protein's primary structure is defined as the amino acid sequence of its polypeptide chain; secondary structure is the local spatial arrangement of a polypeptide's backbone (main chain) atoms; tertiary structure refers to the three-dimensional structure of an entire polypeptide chain; and quaternary structure is the ...
What is an example of protein secondary structure?
The two most important secondary structures of proteins, the alpha helix and the beta sheet, were predicted by the American chemist Linus Pauling in the early 1950s.
What is a major difference between secondary and tertiary structure?
While secondary structure is created solely by hydrogen bonding between the N-H and C=O. groups on the amino acid chain backbone, the tertiary structure is determined by interactions of amino acid R-groups (also known as side chains), with other R-groups and the environment.
How does protein secondary structure play an important role in protein folding?
Secondary structure is generated by formation of hydrogen bonds between atoms in the polypeptide backbone, which folds the chains into either alpha helices or beta-sheets. Tertiary structure is formed by the folding of the secondary structure sheets or helices into one another.
What is the secondary structure of a protein?
The secondary structure of a protein is due to the folding of the polypeptide chain into different folds due to hydrogen bonding and Vander Waal forces. Whereas the tertiary structure of proteins is defined as the arrangement of secondary structure content in 3-dimensional space. While some proteins consist of more than one polypeptide, ...
What are the three basic levels of structure arrangement of a protein?
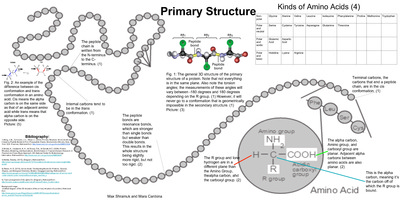
There are three basic levels of structure arrangement of a protein which consist of a single polypeptide, called primary protein structure, secondary protein structure, and tertiary protein structure . The primary protein structure is a simple sequence of the amino acids in which they arrange in a polypeptide chain.
How many amino acids are in the helical structure?
Each turn of the α-helix contains 3.6 amino acids and the helical structure rise along its axis to 5.4 Å.
What are the angles that stabilize the polypeptide chain?
These angles are called torsion angles and help in the folding of the polypeptide chain into different secondary structure elements like α-helix, β-sheet, β pleated-sheet, and turns. These secondary structure elements are also stabilized by the forces present between amino acids located at some distance from each other.
What is the most common helical structure?
There are different types of helical structure were observed in the proteins but the most common is the α-helix. The helical structure forms due to the presence of the turns in the polypeptide chain and different helical structure are identified on the basis ...
What are the forces that form a protein?
These forces are hydrogen bonding and the van der Waal forces. Secondary structure elements present in repetitive forms in a protein and some proteins rich in α-helix content and others in β-sheet while others have mixed ratio of α-helix and β-sheet contents.
What is the proline residue in a protein called?
Mostly, proline residue is present in these turn and they are called β turn . In other cases, polypeptide strands located at different places in a protein can form a hydrogen bond with each other and these are often joined by a long stretch of a polypeptide called loops and sometimes secondary structure like α-helix present in loop regions. The turn of the loop region which joined the two strands can be a right-handed cross over or a left-handed cross over which is rarely present in a β pleated-sheet.
What is the most common form of protein secondary structure?
In structural biology, protein secondary structure is the general three-dimensional form of local segments of proteins. Alpha helices and beta sheets are the most common protein secondary structures.
Does a protein have a glycine?
For explanation, a given protein might have a glycine at a specific position, which by itself might support a random coil there. Whereas, multiple sequence alignment might indicate that helix-favoring amino acids occur at this position (and nearby positions) in 95% of homologous proteins spanning about a billion years of evolution.
How to predict protein structure?
So far it is the most accurate way to predict protein structure by taking its homologous structure in PDB as template. With the rapid growth of PDB database, an increasing proportion of target proteins can be predicted via homology modeling. When no structure with obvious sequence similarity to the target protein can be found in PDB, it is still possible to find out proteins with structural similarity to the target protein.5The method to identify template structures from the PDB is called threading or fold recognition,6–8which actually matches the target sequence to homologous and distant-homologous structures based on some algorithm and take the best matches as structural template. The basic premise for threading to work is that protein structure is highly conservative in evolution and the number of unique structural folds are limited in nature.9,10
What are the steps of protein structure prediction?
Although the detailed prediction processes involved in different prediction methods vary widely, the fundamental steps are consistent, including conformation initialization, conformational search, structure selection, all-atom structure reconstruction and structure refinement (as shown in Fig. 1). In this paper, we will introduce these steps in turn, including some well-known methods or tools commonly used in each step. We will also present a short overview of the critical assessment of protein structure prediction (CASP) experiments13,14and some discussions about the current difficulties and future directions of protein structure prediction.
How are target proteins selected for CASP?
Depending on whether a suitable template can be identified or not, the domains of all target proteins are divided into two basic categories, the template based modeling (TBM) and the template-free modeling (FM). Then pertinent assessments can be implemented on the predicted models of each category. Furthermore, prediction methods are also divided into two categories, those using a combination of computational methods and human experience (Human Section), and those relying solely on computational methods (Server Section). Generally, a window of three weeks is provided for prediction of a target by human-expert groups and three days by servers. After the closing of the server prediction window, the server models are posted at the Prediction Center web site (http://predictioncenter.org), which can be further used by human-expert predictors as starting points for more detailed modeling. Once all models of a target protein have been collected, the Prediction Center performs a standard numerical evaluation of the models by taking the experimental structure as the gold standard. The identity of the predictors is concealed from the assessors when they conduct their analysis. At the predictors’ meeting in December of a CASP year, the results of the evaluations are presented to the community and also made publicly available through the Prediction Center web site. The articles by the organizers, the assessors, and the most successful prediction groups are published in special issues of the journal Proteins: Structure, Function, and Bioinformatics.
What is the key factor in protein structure selection?
Even with the global minimal state identified, the conformation is not necessarily corresponding to the one closest to native state because of the inadequacies of force field. Thus, the common procedure during simulation is to regularly output lower energy intermediate structures for subsequent conformational screening. The key factor of structure selection is the assessment method for distinguishing native-like structures from nonnative ones. There is a specific prediction category in CASP for assessing the methods of structural quality assessment.38It should be noted that the methods for structure selection may be designed specifically for assessing the reduced structural models corresponding to the simplified representation adopted during conformational search. It is an important research direction in protein structure prediction to develop methods of structural quality assessment based on all kinds of ideas and techniques. Furthermore, there are many structural decoys generated by diverse methods39–41available for training and testing methods of structural quality assessment, including the structural models from CASP.
How to identify homologous protein?
In most cases the structural template homologous to the target protein can be identified from the PDB database by sequence alignment,15–17and an accurate alignment between target and template can be built . The similarity between sequences often implies the similarity between their structures. Sometimes the conformation copied from the template (not necessarily complete) is already very close to the optimal one; this is especially the case to homology modeling, where the key issue for homology modeling is thus to improve the efficiency and accuracy of sequence alignment. Since the protein structure is much more conservative than its sequence, it is often the case that two proteins without any sequential similarity fold into similar folds, which is actually the foundation of threading methods. During the past two decades, the threading method has been widely studied and applied, which greatly improves the utilization of the experimentally solved structures. In general, the initial conformation obtained from structural template is vastly better than any ones built from scratch and can dramatically shorten the process of subsequent conformational search.
How to identify structural template?
For most target proteins, the desirable structural template can be identified from PDB by sequence alignment or threading method. Since the conformational information from template is much more reliable than that from elsewhere (especially when the target protein and the template are highly homologous), the prediction accuracy of template-based method is generally higher than other methods, which makes it highly popular in practical applications.
How do proteins differ from each other?
Proteins differ from one another primarily in their sequence of amino acids, which usually results in different spatial shape and structure and therefore different biological functionalities in cells. However, so for little is known about how protein folds into the specific three-dimensional structure from its one-dimensional sequence. In comparison with the genetic code by which a triple-nucleotide codon in a nucleic acid sequence specifies a single amino acid in protein sequence, the relationship between protein sequence and its steric structure is called the second genetic code (or folding code).1
Why is protein structure prediction important?
Protein structure prediction is one of the most important goals pursued by bioinformatics and theoretical chemistry; it is highly important in medicine (for example, in drug design) and biotechnology (for example, in the design of novel enzymes). protein structure prediction introduction from: ...
What can be analyzed to predict secondary, tertiary and quaternary protein structure?
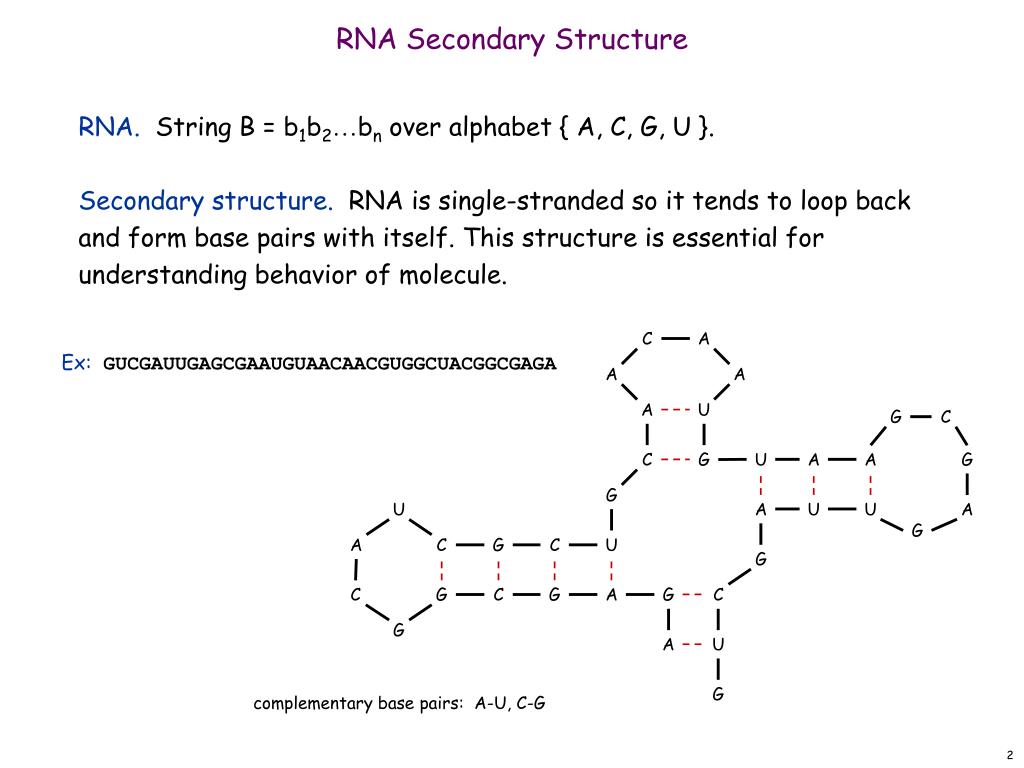
Constituent amino-acids can be analyzed to predict secondary, tertiary and quaternary protein structure. ( Picture from wiki)
What are the three methods of predicting the structure of proteins?
There are three major theoretical methods for predicting the structure of proteins: comparative modelling, fold recognition , and ab initio prediction.
What is secondary structure prediction?
Protein secondary structure prediction refers to the prediction of the conformational state of each amino acid residue of a protein sequence as one of the three possible states, namely, helices, strands, or coils, denoted as H, E, and C, respectively. The prediction is based on the fact that secondary structures have a regular arrangement of amino acids, stabilized by hydrogen ..
What is the prediction of the conformational state of each amino acid residue of a protein sequence?
The prediction is based on the fact that secondary structures have a regular arrangement of amino acids, stabilized by hydrogen .. Protein secondary structure prediction refers to the prediction of the conformational state of each amino acid residue of a protein sequence as one of the three possible states, namely, helices, strands, or coils, ...
How to calculate propensity score?
The calculation of residue propensity scores is simple. Suppose there are n residues in all known protein structures from which m residues are helical residues. The total number of alanine residues is y of which x are in helices. The propensity for alanine to be in helix is the ratio of the proportion of alanine in helices over the proportion of alanine in overall residue population (using the formula [ x/m] / [ y/n ]). If the propensity for the residue equals 1.0 for helices ( P [ α -helix]), it means that the residue has an equal chance of being found in helices or elsewhere. If the propensity ratio is less than 1, it indicates that the residue has less chance of being found in helices. If the propensity is larger than 1, the residue is more favoured by helices. Based on this concept, Chou and Fasman developed a scoring table (Secondary structure element score) listing relative propensities of each amino acid to be in an α -helix, a β -strand, or a β -turn.
What does it mean when the propensity ratio is less than 1?
If the propensity ratio is less than 1, it indicates that the residue has less chance of being found in helices. If the propensity is larger than 1, the residue is more favoured by helices. ...
What is ab initio prediction?
This type of method predicts the secondary structure based on a single query sequence. It measures the relative propensity of each amino acid belonging to a certain secondary structure element. The propensity scores are derived from known crystal structures. Examples of ab initio prediction are the Chou–Fasman and Garnier, Osguthorpe, Robson (GOR) methods. The ab initio methods were developed in the 1970s when protein structural data were very limited. The statistics derived from the limited data sets can therefore be rather inaccurate. The methods are simple and are often used to illustrate the basics of secondary structure prediction.
What is a homology based method?
The homology-based methods do not rely on statistics of residues of a single sequence, but on common secondary structural patterns conserved among multiple homologous sequences. This type of method predicts the secondary structure based on a single query sequence.
Why is secondary structure important?
Secondary structures are much more conserved than sequences during evolution. As a result, correctly identifying secondary structure elements (SSE) can help to guide sequence alignment or improve existing sequence alignment of distantly related sequences. Secondary structure prediction is an intermediate step in tertiary structure prediction as in ...
Why run a secondary structure prediction on a newly determined sequence?
Running a secondary structure prediction on a newly-determined sequence just because everyone else does so, is to be deplored, and the fact that the results of such predictions are generally ignored is insufficient justification for doing and publishing them.". Arthur Lesk, 1988.
What is PredictProtein 2013?
PredictProtein 2013(Technical University of Berlin, Germany)- they have substantially expanded the breadth of structural annotations, e.g. by adding predictions of non-regular secondary structure and intrinsically disordered regions, disulphide bridges and inter-residue contacts, and finally by also covering trans-membrane beta barrels structures. They have also added important resources for the prediction of protein function. registration required.
What is proteus2 web server?
PROTEUS2- is a web server designed to support comprehensive protein structure prediction and structure-based annotation. PROTEUS2 accepts either single sequences (for directed studies) or multiple sequences (for whole proteome annotation) and predicts the secondary and, if possible, tertiary structure of the query protein(s). Unlike most other tools or servers, PROTEUS2 bundles signal peptide identification, transmembrane helix prediction, transmembrane β-strand prediction, secondary structure prediction (for soluble proteins) and homology modeling (i.e. 3D structure generation) into a single prediction pipeline. (Reference: Montgomerie S et al. (2008) Nucleic Acids Res. 36(Web Server issue):W202-209).
What is TMRPres2D?
TMRPres2D(TransMembrane protein Re-Presentation in 2Dimensions tool) - this Java tool takes data from a variety of protein folding servers and creates uniform, two-dimensional, high analysis graphical images/ models of alpha-helical or beta-barrel transmembrane proteins. (Reference:I.C. Spyropoulos et al. 2004. Bioinformatics 20:3258-3260).
What is the PRED TMR2?
PRED-TMR2 (C. Pasquier & S.J.Hamodrakas,Dept. Cell Biology and Biophysics, Univ. Athens, Greece)- when applied to several test sets of transmembrane proteins the system gives a perfect prediction rating of 100% by classifying all the sequences in the transmembrane class. Only 2.5% error rate with nontransmembrane proteins.
What is a phobic predictor?
Phobius- is a combined transmembrane topology and signal peptide predictor (Reference:L. Käll et al. 2004. J. Mol. Biol. 338:1027-1036) This tool can also be accessed here (EBI).
Why are globular proteins not stable?
IUPred- The underlying assumption is that globular proteins are composed of amino acids which have the potential to form a large number of favorable interactions, whereas intrinsically unstructured proteins (IUPs) adopt no stable structure because their amino acid composition does not allow sufficient favorable interactions to form. (Reference:Z. Dosztányi et al. 2005. Bioinformatics 21:3433-3434).