
What are some interesting facts about sickle cell anemia?
Surprising Facts About Sickle Cell Anemia
- One Of The Most Common Genetic Disorders. Sickle cell anemia is one of the most common genetic disorders in the world today. ...
- It Can Occur In Any Ethnic Group. ...
- Sickle Cell And Malaria. ...
- It’s More Than Just Pain. ...
- Life Expectancy Has Improved. ...
- There Is A Cure For It. ...
- It’s Not Contagious. ...
What happens in sickle cell anemia, exactly?
Symptoms of sickle cell anemia often start when a baby is a few months old and may include:
- Pain crisis, also called sickle crisis In the bones, chest, or other parts of the body May be mild or severe Can last hours to days In babies Pain in ...
- Severe anemia Tiredness/ fatigue Weakness Dizziness Shortness of breath
- Severe and sometimes life-threatening infections
What race is most affected by sickle cell anemia?
What race is most affected by sickle cell anemia? People of African descent, including African-Americans (among whom 1 in 12 carries a sickle cell gene) Hispanic-Americans from Central and South America. People of Middle Eastern, Asian, Indian, and Mediterranean descent.
Is sickle cell anemia a fatal disease?
Sickle cell disease is the name for a group of inherited health conditions that affect the red blood cells. The most serious type is called sickle cell anaemia. ... It can lead to health problems like strokes, serious infections and lung problems, which can occasionally be fatal. Overall, the life expectancy for someone with sickle cell disease ...

What is the pathophysiology behind sickle cell anaemia?
Sickle cell disease is caused by a mutation in the beta-globin chain of the haemoglobin molecule. Sickle haemoglobin, the result of this mutation, has the singular property of polymerizing when deoxygenated. Exactly how normal tissue perfusion is interrupted by abnormal sickle cells is complex and poorly understood.
What is pathophysiology of vaso-occlusive crisis?
Vaso-occlusive crisis results from the sickle red cells obstructing and reducing blood flow to the vital organs leading to ischemia, necrosis and pain. Repeated episodes lead to bone infarction and necrosis; and bone marrow degeneration occurs overtime.
How does sickle cell anemia affect blood flow?
These damaged red blood cells (sickle cells) clump together. They can't move easily through the blood vessels. They get stuck in small blood vessels and block blood flow. This blockage stops the movement of healthy oxygen-rich blood.
What are the three types of crisis in sickle cell Anaemia?
Four major types of crises are recognised in sickle cell anaemia: aplastic, acute sequestration, hyper-haemolytic, and vaso-occlusive crises.
What happens during vaso-occlusive crisis?
What is a vaso-occlusive crisis? A vaso-occlusive crisis, or VOC, occurs when sickled red blood cells block blood flow to the point that tissues become deprived of oxygen. This in turn sets in motion an inflammatory response as the body tries to rectify the problem.
How does infection cause vaso-occlusive crisis?
Increased oxygen consumption by activated neutrophils lead to increased production of deoxy-HbS. Activated neutrophils generate more free radicals, decrease antioxidants and predispose to red cell sickling. Increased adhesion of neutrophils to sickled cells and vascular endothelium lead to vascular occlusion.
What is the cause of a sickle cell patient's distress when they are in vaso-occlusive crisis?
Polymerization of deoxygenated sickle hemoglobin leads to decreased deformability of red blood cells (RBCs). Through a complex interplay of adhesive events among blood cells, these altered erythrocytes can obstruct the vasculature, producing episodes of pain, hemolytic anemia, organ injury, and early mortality.
What are the manifestations of vaso-occlusive crisis?
A vaso-occlusive crisis most commonly involves the back, legs, knees, arms, chest and abdomen. The pain generally affects two or more sites. Bone pain tends to be bilateral and symmetric. Recurrent crises in an individual patient usually have the same distribution.
What is the first sign of sickle cell anemia?
Fever. People with sickle cell anemia have an increased risk of serious infection, and fever can be the first sign of an infection.
Why do doctors give sickle cell anemia?
Doctors commonly give infants and children with sickle cell anemia vaccinations and antibiotics to prevent potentially life-threatening infections, such as pneumonia. Delayed growth or puberty. Red blood cells provide your body with the oxygen and nutrients needed for growth.
What does sickle cell anemia look like?
Overview. Normal red blood cells are rounded and disk-shaped. In sickle cell anemia, some red blood cells become deformed, so they look like sickles used to cut wheat. These unusually shaped cells give the disease its name. Sickle cell anemia is one of a group of disorders known as sickle cell disease.
What happens if you have sickle cells in your eyes?
Tiny blood vessels that supply your eyes can become plugged with sickle cells. This can damage the retina — the portion of the eye that processes visual images — and lead to vision problems.
How long do sickle cells last?
Red blood cells usually live for about 120 days before they need to be replaced. But sickle cells usually die in 10 to 20 days, leaving a shortage of red blood cells (anemia).
What is the name of the substance that is produced by the breakdown of red blood cells?
Gallstones. The breakdown of red blood cells produces a substance called bilirubin. A high level of bilirubin in your body can lead to gallstones.
Can anemia cause a stroke?
Sickle cell anemia can lead to a host of complications, including: Stroke. Sickle cells can block blood flow to an area of your brain. Signs of stroke include seizures, weakness or numbness of your arms and legs, sudden speech difficulties, and loss of consciousness.
What is the cause of sickle cell disease?
Sickle cell disease is caused by a mutation in the beta-globin chain of the haemoglobin molecule. Sickle haemoglobin, the result of this mutation, has the singular property of polymerizing when deoxygenated.
What is the singular property of sickle haemoglobin?
Sickle haemoglobin, the result of this mutation, has the singular property of polymerizing when deoxygenated. Exactly how normal tissue perfusion is interrupted by abnormal sickle cells is complex and poorly understood.
What is the pathophysiology of sickle cell anemia?
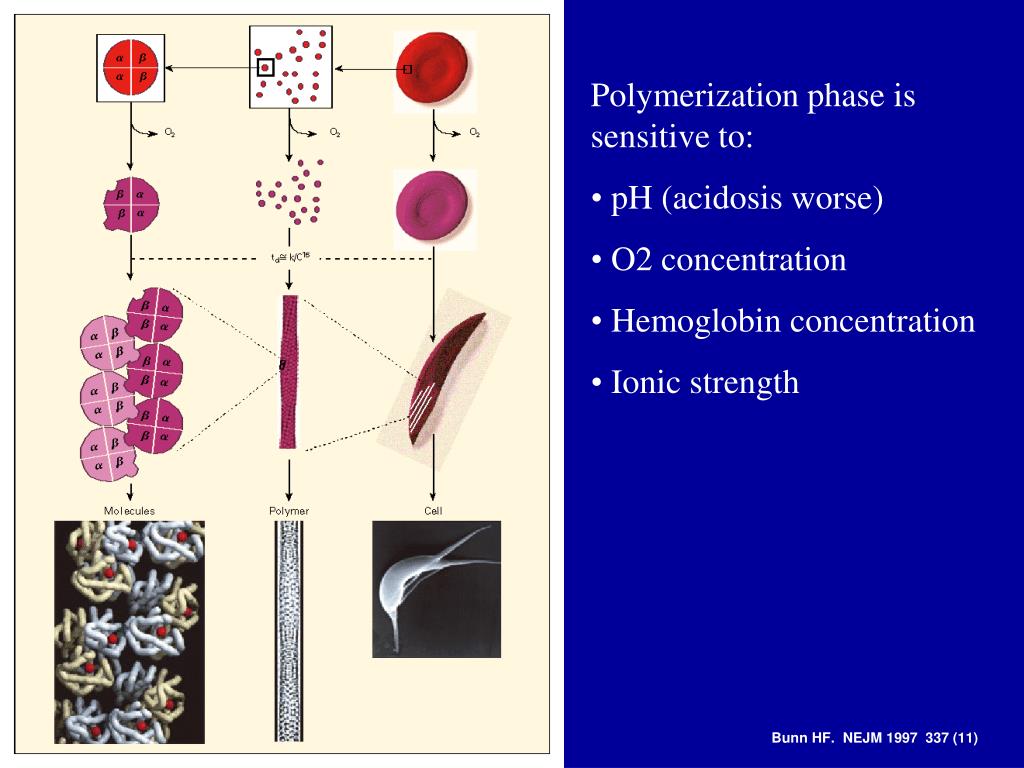
Schematic representation of the pathophysiology (in part) of sickle cell anemia. A single gene mutation (GAG→GT G and CTC→CAC) results in a defective haemoglobin that when exposed to de-oxygenation (depicted in the right half of the diagram) polymerizes (upper right of the diagram), resulting in the formation of sickle cells. Vaso-occlusion can then occur. The disorder is also characterized by abnormal adhesive properties of sickle cells; peripheral blood mononuclear cells (depicted in light blue; shown as the large cells under the sickle cells) and platelets (depicted in dark blue; shown as the dark circular shapes on the mononuclear cells) adhere to the sickled erythrocytes. This aggregate is labelled 1. The mononuclear cells have receptors (e.g., CD44 (labeled 3 and depicted in dark green on the cell surface)) that bind to ligands, such as P-selectin (labeled 2 and shown on the endothelial surface), that are unregulated. The sickle erythrocytes can also adhere directly to the endothelium. Abnormal movement or rolling and slowing of cells in the blood also can occur. These changes result in endothelial damage. The sickled red cells also become dehydrated as a result of abnormalities in the Gardos channel. Hemolysis contributes to oxidative stress and dysregulation of arginine metabolism, both of which lead to a decrease in nitric oxide (NO) that, in turn, contributes to the vasculopathy that characterizes SCD.
What is sickle cell disease?
Sickle cell disease (SCD) is a monogenetic disorder due to a single base-pair point mutation in the β-globin gene resulting in the substitution of the amino acid valine for glutamic acid in the β-globin chain. Phenotypic variation in the clinical presentation and disease outcome is a characteristic feature of the disorder. Understanding the pathogenesis and pathophysiology of the disorder is central to the choice of therapeutic development and intervention. In this special edition for newborn screening for haemoglobin disorders, it is pertinent to describe the genetic, pathologic and clinical presentation of sickle cell disease as a prelude to the justification for screening. Through a systematic review of the literature using search terms relating to SCD up till 2019, we identified relevant descriptive publications for inclusion. The scope of this review is mainly an overview of the clinical features of pain, the cardinal symptom in SCD, which present following the drop in foetal haemoglobin as young as five to six months after birth. The relative impact of haemolysis and small-vessel occlusive pathology remains controversial, a combination of features probably contribute to the different pathologies. We also provide an overview of emerging therapies in SCD.
What mutations cause sickle cell polymerisation?
The sickle β-globin mutation renders the sickle gene pleiotropic in nature, with variable phenotypic expression associated with complex genetic and environmental interactions, as well as disease modifiers that are increasingly being recognised. Sickle RBC (sRBC) polymerisation in deoxygenated environments is influenced by a number of factors including the co-inheritance of alpha thalassaemia, foetal haemoglobin (HbF) level which is determined by a number of genetic factors including genetic variations of BLC11A, HBS1L-MYB and HBB loci and hydroxycarbamide therapy amongst others [35]. The BCL11A and ZBTB7A genes (LRF protein) are responsible for the suppression of γ chains, and HbF production. HbF reduces sickle cell polymerisation due to reduction in HbS concentration and the fact that it is excluded from the sickle cell polymers. HbF also has high oxygen retention thereby ameliorating both the vaso-occlusive and haemolytic pathology in SCD. Among the four main sickle haplotypes, the level of HbF is highest in the Indian/Arab haplotype [8] predominantly found in the Arab Peninsula and India followed by the Senegal, and Benin haplotypes most predominant in Sub-Saharan Africa. The Bantu haplotype with the lowest HbF is predominantly found in Central African countries [8,36]. Alpha thalassaemia also modulates the expression of SCD. People with one or two α genes deleted have less haemolysis and fewer vasculopathy complications [8].
What is SCD in biology?
Sickle cell disease (SCD) was first reported by Herrick in 1910 even though reports suggest prior description of the disorder [1]; it is the result of homozygous and compound heterozygote inheritance of a mutation in the β-globingene. A single base-pair point mutation (GAG to GTG) results in the substitution of the amino acid glutamic acid (hydrophilic) to Valine (hydrophobic) in the 6th position of the β-chain of haemoglobin referred to as haemoglobin S (HbS) [2]. Phenotypic variation in clinical presentation is a unique feature of SCD despite a well-defined Mendelian inheritance, the first to be molecularly characterised as described by Pauling [3] and confirmed to be due to a single amino acid substitution by Ingram [3] almost 70 years ago. SCD is a multi-organ, multi-system disorder with both acute and chronic complications presenting when foetal haemoglobin (HbF) drops towards the adult level by five to six months of age [4].
Why do people with SCD need blood transfusions?
Individuals with SCD have a baseline level of anaemia due to their chronic haemolysis. Blood transfusions are not given to correct this baseline anaemia or for acute pain episodes. Instead, transfusions are given to correct acute severe anaemia where the haemoglobin falls significantly below that individual’s baseline, and the resulting impairment in oxygen delivery to body tissues would otherwise propagate further sickling of deoxygenated Hb. Examples include red cell aplasia caused by Parvovirus B19 infection, acute splenic sequestration or hyperhaemolysis crises [71]. In an acute setting, transfusion is also used to bridge periods of severe physiologic stress like major surgery or critical illness including acute chest crises. In this setting, blood transfusion with HbS-negative blood reduces the proportion of circulating haemoglobin that is able to sickle, and hence reducing vessel occlusion and haemolysis from abnormal sickle RBCs. Long-term transfusions are instituted as a disease-modifying treatment in specific situations, such as to prevent stroke. The issues that arise as a result of long term transfusion complications include: (i) Allo-immunization, where after receiving a blood transfusion an individual develops antibodies to an antigen on the transfused red cells, which can increase the risk of having haemolytic reactions to blood that they are transfused in future; (ii) Iron overload, although treatment of iron overload is becoming more tolerable with the new oral chelators and, (iii) Risk of transfusion-transmitted infections, especially in countries where only limited screening of donated blood is available [39,72].
What causes sickle cell acute chest syndrome?
SCD increases susceptibility to infections, notably bacterial sepsis and malaria in children under five years [45]. Respiratory infections can trigger the sickle-cell acute chest syndrome, with a high risk of death. Risk factors for infections include: (i) functional asplenia/hyposplenia which present with reduced splenic immune response at a very young age, (ii) impaired fixation of complement, (iii) reduced oxidative burst capacity of chronically activated neutrophils, dysfunctional IgM and IgG antibody responses and defective opsonisation. The main pathogen of concern is Streptococcus pneumoniae,though severe and systemic infections arise with Haemophilus influenzae, Neisseria meningitides, and Salmonellaeleads to osteomyelitis especially Salmonelladue to bowel ischaemia and gut flora dissemination [46,47].
What causes bone marrow failure in SCD?
The most common cause of acquired bone marrow failure in SCD and other haemolytic disorders is caused by Parvovirus B19 [44]. This virus causes the fifth disease and normally in healthy children is quite mild, associated with malaise, fever and sometimes a mild rash; the virus affects erythropoiesis by invading progenitors of RBCs in the bone marrow and destroying them, thus preventing new RBCs from being made. There’s a slight drop in haematocrit in children with HbAA who are generally unaffected, however in SCD as the lifespan of RBC’s is reduced to about 10–20 days, there is a significant drop in haemoglobin concentration. Parvovirus B19 infection usually takes about four days to one week to resolve and patients with SCD usually require a blood transfusion [39,44].
Why do sickle cells cause pain?
Sickle cell crises may result due to the increased viscosity of the blood and the formation of blockages in the blood vessels. When the rigid cells group together, they can disrupt the flow of oxygen and restrict the supply to tissues that require oxygenation. This results in sudden and severe pain, known as sickle cell crisis, which usually requires medical management.
Why do cells stay in a sickle shape?
Over time, the membrane of the cells becomes permanently damaged, leading cells to permanently stay in the bi-concave sickle shape, even when the blood is exposed to sufficient levels of oxygen once again .
What is the condition that causes a blockage in the blood vessels?
Sickle cell disease is an inherited genetic condition that involves defects in the shape and function of hemoglobin in the blood. This increases the likelihood of blockages in the blood vessels and disrupted blood flow, which can result in serious complications. Image Credit: Kateryna Kon / Shutterstock.com.
What happens when a cell is shaped?
As a result, the cells are more likely to hemolyze and cause blockages in the blood vessels that disrupt the flow of blood.
What is the chance of a sickle cell?
When both parents carry a single gene mutation, known as the sickle cell trait, there is a 25% chance that the disease will develop in their offspring, a 25% chance that the child will be unaffected and a 50% that they will possess the genetic mutation as an asymptomatic carrier.
What is the role of hemoglobin in the body?
Hemoglobin is an essential cellular component of the red blood cells that play the role of transporting oxygen in the blood to the bodily tissues where it is required. Sickle hemoglobin differs in physical shape from normal hemoglobin, with a curved sickle-shape rather than flat-disc-shaped cells.
What happens to hemoglobin when there is no oxygen?
When there is insufficient oxygen in the vascular system, sickle hemoglobin becomes considerably more insoluble, increasing the polymer formation in the blood and its overall viscosity. This leads to the formation of tactoids, which are a gel-like form of hemoglobin, that exists in equilibrium with its ordinary soluble state.
How is the HBB gene mutated in sickle cell disease?
Genes are made up of a string of pieces called “nucleotides.” The body’s cells use the specific order of nucleotides as a template to make a specific protein. Proteins are long strings of fragments called “amino acids.” Mutation of a single nucleotide on the HBB gene causes a single amino acid mutation on the beta-globin protein. This is an example of a “missense mutation.”
How many sickle beta globins are in the human body?
It has 2 normal alpha globins but contains 2 sickle beta globins. Sickle beta-globin has an amino acid called “valine” instead of an amino acid called “glutamic acid” at a specific location in the protein. Other forms of abnormal hemoglobin are caused by different mutations in the HBB gene. 1.
How is the mutated HBB gene inherited?
We have 2 copies of 23 chromosomes, with 1 copy coming from each parent. This means we inherit 1 copy of every gene from each parent. However, mutations may cause each copy to have a slightly different nucleotide sequence. Different variants of the same gene are called “alleles.”
What causes hemoglobin to cluster?
The mutation in beta-globin causes hemoglobin to cluster together and misshape the red blood cells. This leads to the symptoms and complications experienced by people with SCD. Blood tests can identify HBB mutations and abnormal hemoglobin. 3
What is SCD in biology?
By Editorial Team. October 28, 2020. Sickle cell disease (SCD) is a genetic disorder caused by a mutation in the HBB gene. This gene provides instructions for the body to produce a part of hemoglobin. Hemoglobin is a protein that carries oxygen throughout the body.
What is the chance of a child having sickle cell?
There is a 50 percent chance the child will have sickle cell trait. There is a 25 percent chance the child will not have either condition. If 1 parent has SCD and 1 parent has sickle cell trait. There is a 50 percent chance the child will have SCD. There is a 50 percent chance the child will have sickle cell trait. If both parents have SCD.
Can SCD be inherited?
This can lead to anemia and blocked blood flow for people with SCD. SCD can only be inherited if each parent passes down a mutated HBB gene. This means it is a recessive trait.
Overview
Symptoms
- Signs and symptoms of sickle cell anemia usually appear around 6 months of age. They vary from person to person and may change over time. Signs and symptoms can include: 1. Anemia.Sickle cells break apart easily and die. Red blood cells usually live for about 120 days before they need to be replaced. But sickle cells typically die in 10 to 20 days, leaving a shortage of red blood cells (a…
Causes
- Sickle cell anemia is caused by a change in the gene that tells the body to make the iron-rich compound in red blood cells called hemoglobin. Hemoglobin enables red blood cells to carry oxygen from the lungs throughout the body. The hemoglobin associated with sickle cell anemia causes red blood cells to become rigid, sticky and misshapen. For a chi...
Risk Factors
- For a baby to be born with sickle cell anemia, both parents must carry a sickle cell gene. In the United States, sickle cell anemia most commonly affects people of African, Mediterranean and Middle Eastern descent.
Complications
- Sickle cell anemia can lead to a host of complications, including: 1. Stroke.Sickle cells can block blood flow to an area of the brain. Signs of stroke include seizures, weakness or numbness of the arms and legs, sudden speech difficulties, and loss of consciousness. If your child has any of these signs and symptoms, seek medical treatment immediately. A stroke can be fatal. 2. Acute …
Prevention
- If you carry the sickle cell trait, seeing a genetic counselor before trying to conceive can help you understand your risk of having a child with sickle cell anemia. A genetic counselor can also explain possible treatments, preventive measures and reproductive options.